

Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health
- PMID: 26100894
- PMCID: PMC4500264
- DOI: 10.1073/pnas.1501049112
Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health
Abstract
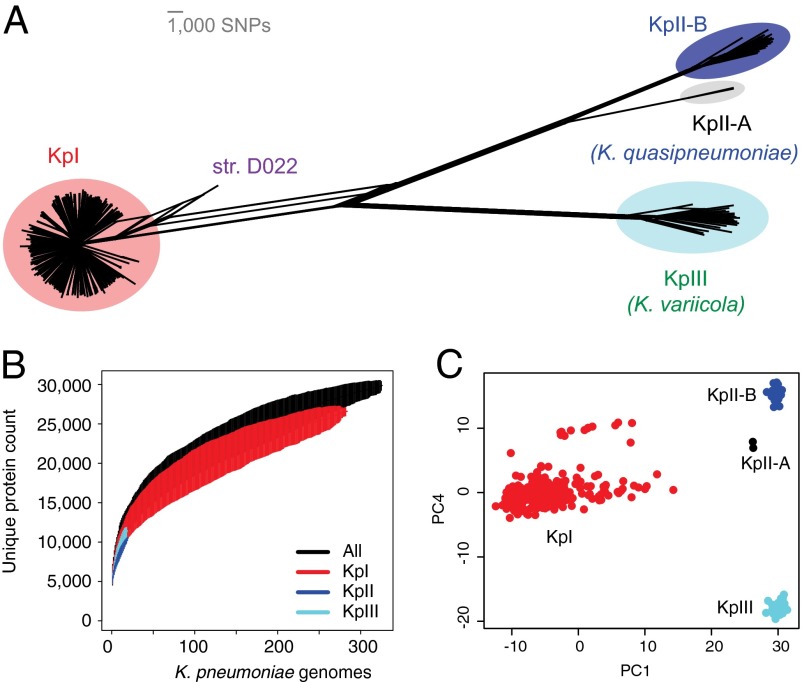
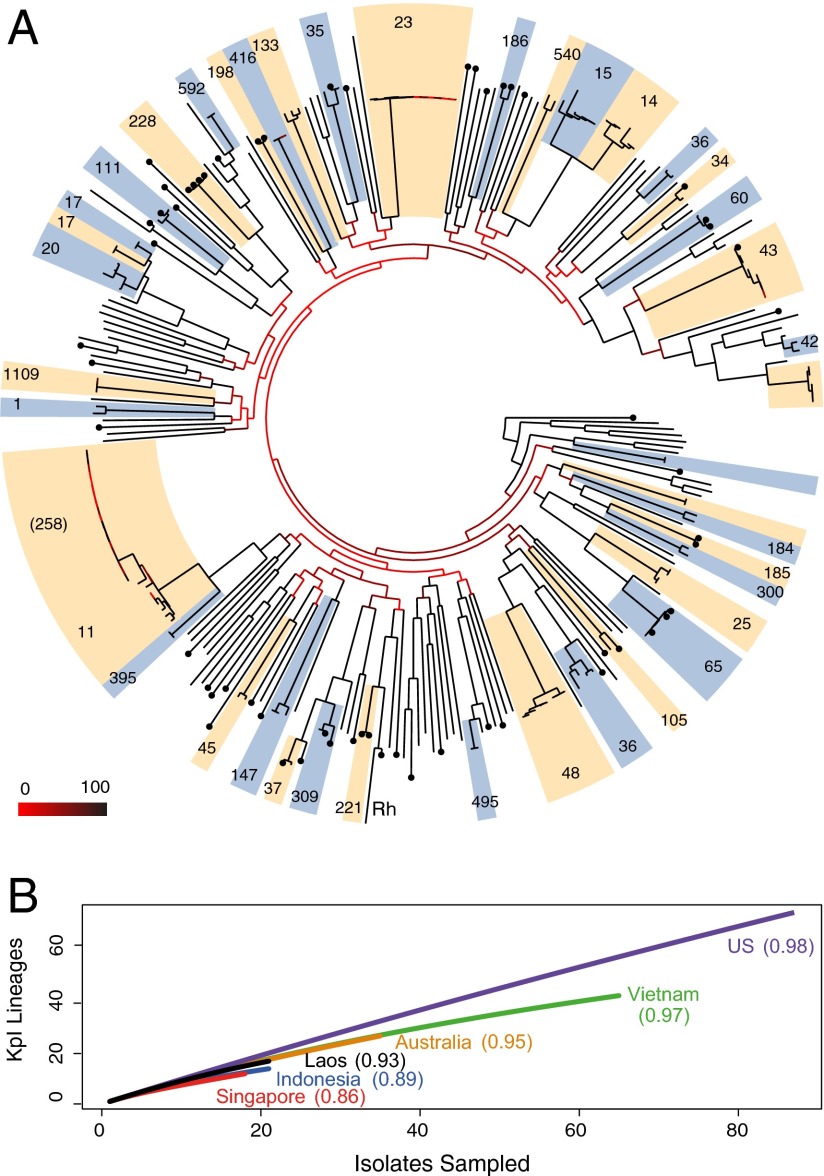
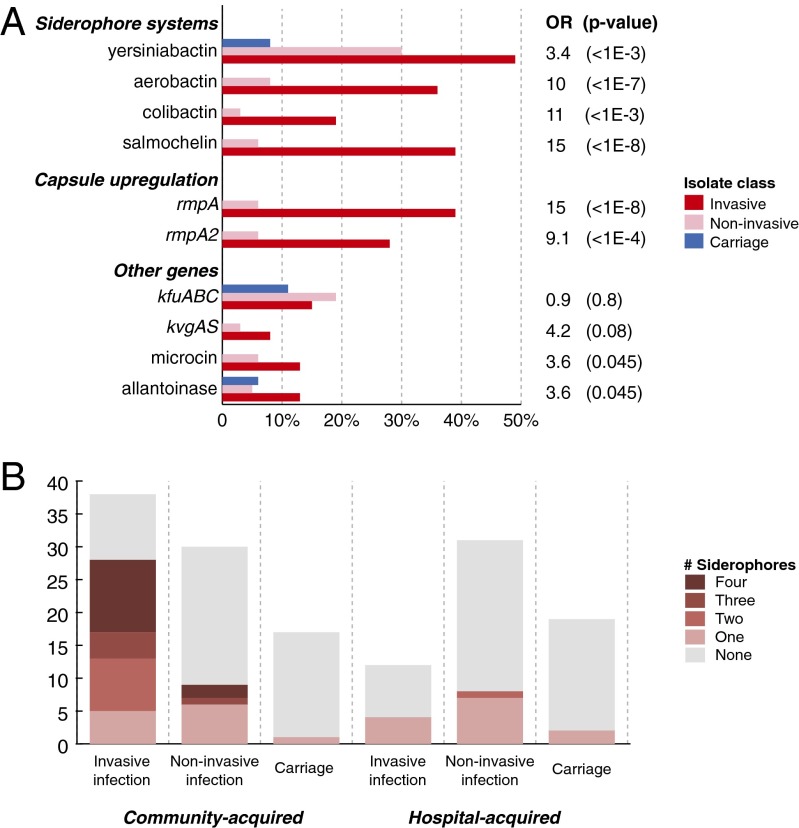
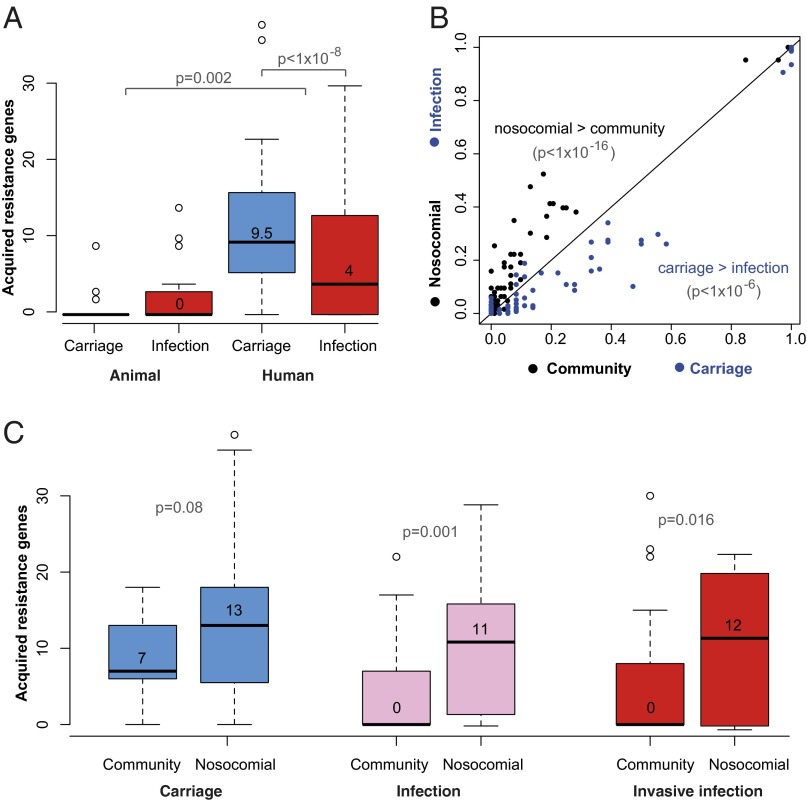
Klebsiella pneumoniae is now recognized as an urgent threat to human health because of the emergence of multidrug-resistant strains associated with hospital outbreaks and hypervirulent strains associated with severe community-acquired infections. K. pneumoniae is ubiquitous in the environment and can colonize and infect both plants and animals. However, little is known about the population structure of K. pneumoniae, so it is difficult to recognize or understand the emergence of clinically important clones within this highly genetically diverse species. Here we present a detailed genomic framework for K. pneumoniae based on whole-genome sequencing of more than 300 human and animal isolates spanning four continents. Our data provide genome-wide support for the splitting of K. pneumoniae into three distinct species, KpI (K. pneumoniae), KpII (K. quasipneumoniae), and KpIII (K. variicola). Further, for K. pneumoniae (KpI), the entity most frequently associated with human infection, we show the existence of >150 deeply branching lineages including numerous multidrug-resistant or hypervirulent clones. We show K. pneumoniae has a large accessory genome approaching 30,000 protein-coding genes, including a number of virulence functions that are significantly associated with invasive community-acquired disease in humans. In our dataset, antimicrobial resistance genes were common among human carriage isolates and hospital-acquired infections, which generally lacked the genes associated with invasive disease. The convergence of virulence and resistance genes potentially could lead to the emergence of untreatable invasive K. pneumoniae infections; our data provide the whole-genome framework against which to track the emergence of such threats.
Keywords: Klebsiella pneumoniae; antimicrobial resistance; genomics; population structure; virulence.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Jones RN. Microbial etiologies of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin Infect Dis. 2010;51(Suppl 1):S81–S87. - PubMed
-
- Falade AG, Ayede AI. Epidemiology, aetiology and management of childhood acute community-acquired pneumonia in developing countries—a review. Afr J Med Med Sci. 2011;40(4):293–308. - PubMed
-
- Jarvis WR, Munn VP, Highsmith AK, Culver DH, Hughes JM. The epidemiology of nosocomial infections caused by Klebsiella pneumoniae. Infect Control. 1985;6(2):68–74. - PubMed
-
- Brisse S, Grimont F, Grimont P. 2006. The genus Klebsiella. The Prokaryotes A Handbook on the Biology of Bacteria, 3rd edition, eds Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (Springer, New York), 3rd Ed. Vol 6: Proteobacteria: Gamma Subclass.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
