

Melanoma patient derived xenografts acquire distinct Vemurafenib resistance mechanisms
- PMID: 26101714
- PMCID: PMC4473327
Melanoma patient derived xenografts acquire distinct Vemurafenib resistance mechanisms
Abstract
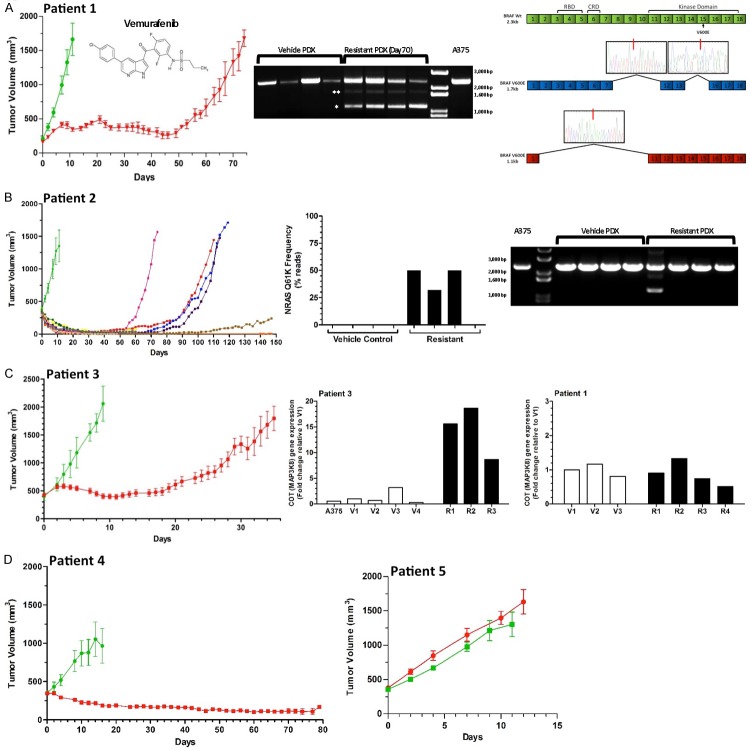
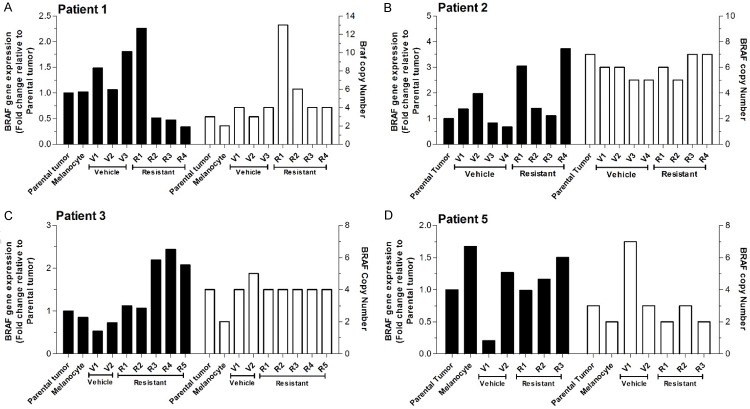
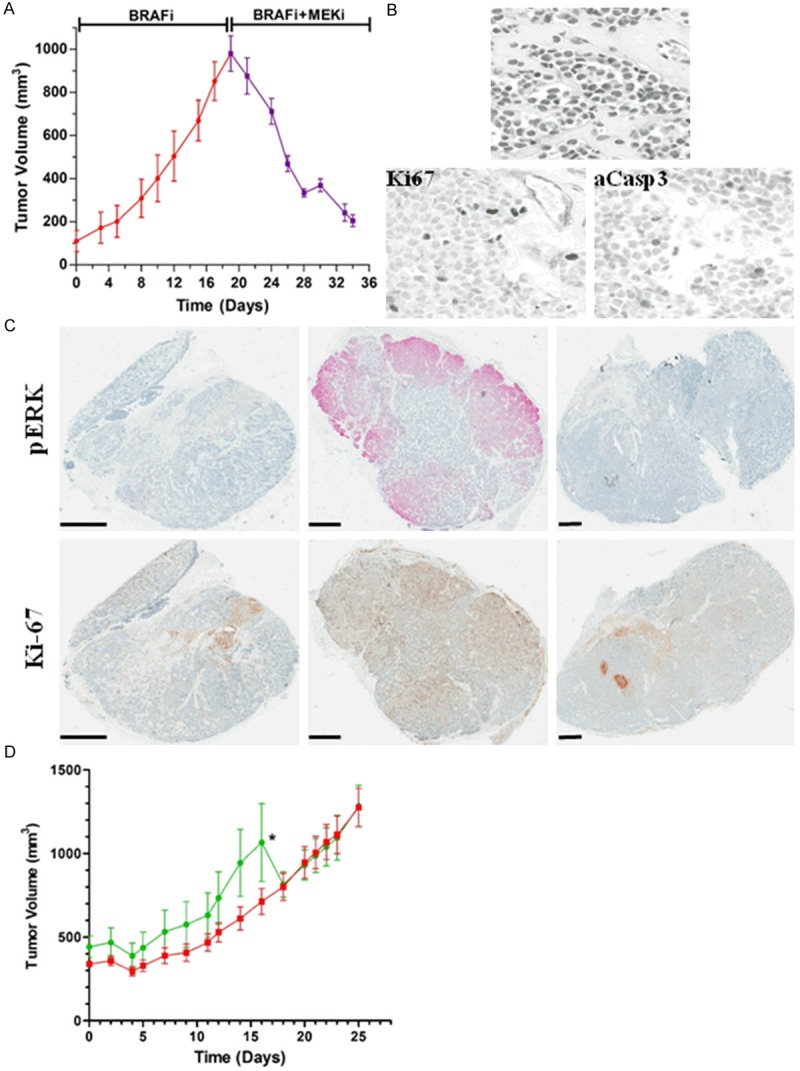
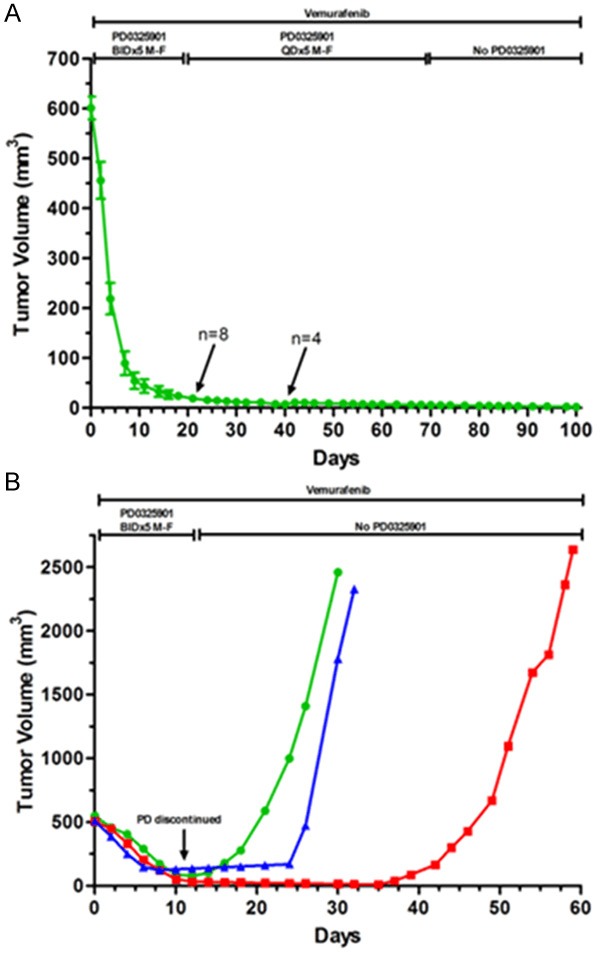
Variable clinical responses, tumor heterogeneity, and drug resistance reduce long-term survival outcomes for metastatic melanoma patients. To guide and accelerate drug development, we characterized tumor responses for five melanoma patient derived xenograft models treated with Vemurafenib. Three BRAF(V600E) models showed acquired drug resistance, one BRAF(V600E) model had a complete and durable response, and a BRAF(V600V) model was expectedly unresponsive. In progressing tumors, a variety of resistance mechanisms to BRAF inhibition were uncovered, including mutant BRAF alternative splicing, NRAS mutation, COT (MAP3K8) overexpression, and increased mutant BRAF gene amplification and copy number. The resistance mechanisms among the patient derived xenograft models were similar to the resistance pathways identified in clinical specimens from patients progressing on BRAF inhibitor therapy. In addition, there was both inter- and intra-patient heterogeneity in resistance mechanisms, accompanied by heterogeneous pERK expression immunostaining profiles. MEK monotherapy of Vemurafenib-resistant tumors caused toxicity and acquired drug resistance. However, tumors were eradicated when Vemurafenib was combined the MEK inhibitor. The diversity of drug responses among the xenograft models; the distinct mechanisms of resistance; and the ability to overcome resistance by the addition of a MEK inhibitor provide a scheduling rationale for clinical trials of next-generation drug combinations.
Keywords: BRAF; MEK; Melanoma; Vemurafenib; drug resistance; patient derived xenografts (PDX).
Figures
References
-
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA. Improved survival with Vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. - PMC - PubMed
-
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WHJ, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, Chiarion-Sileni V, Martin AM, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, Chapman PB. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. - PubMed
-
- Sullivan RJ, Flaherty KT. Major therapeutic developments and current challenges in advanced melanoma. Br J Dermatol. 2014;170:36–44. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous