

Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts
- PMID: 26105199
- PMCID: PMC4568946
- DOI: 10.15252/emmm.201404914
Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts
Abstract
The development of targeted inhibitors, like vemurafenib, has greatly improved the clinical outcome of BRAF(V600E) metastatic melanoma. However, resistance to such compounds represents a formidable problem. Using whole-exome sequencing and functional analyses, we have investigated the nature and pleiotropy of vemurafenib resistance in a melanoma patient carrying multiple drug-resistant metastases. Resistance was caused by a plethora of mechanisms, all of which reactivated the MAPK pathway. In addition to three independent amplifications and an aberrant form of BRAF(V600E), we identified a new activating insertion in MEK1. This MEK1(T55delins) (RT) mutation could be traced back to a fraction of the pre-treatment lesion and not only provided protection against vemurafenib but also promoted local invasion of transplanted melanomas. Analysis of patient-derived xenografts (PDX) from therapy-refractory metastases revealed that multiple resistance mechanisms were present within one metastasis. This heterogeneity, both inter- and intra-tumorally, caused an incomplete capture in the PDX of the resistance mechanisms observed in the patient. In conclusion, vemurafenib resistance in a single patient can be established through distinct events, which may be preexisting. Furthermore, our results indicate that PDX may not harbor the full genetic heterogeneity seen in the patient's melanoma.
Keywords: Melanoma; drug resistance; patient‐derived xenografts; tumor heterogeneity.
© 2015 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
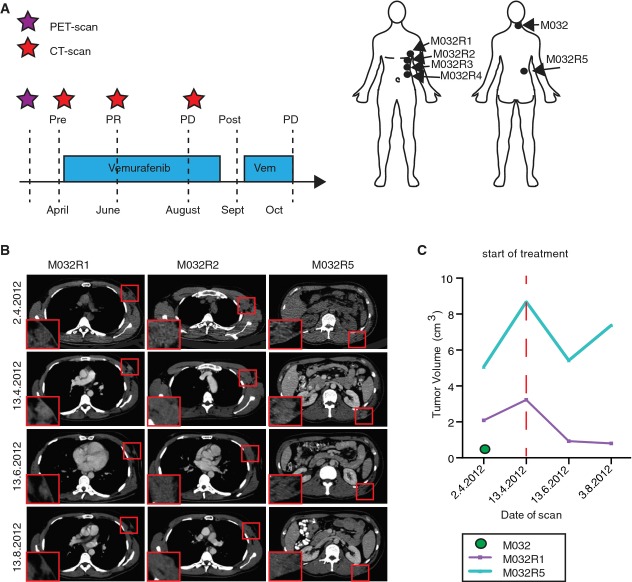
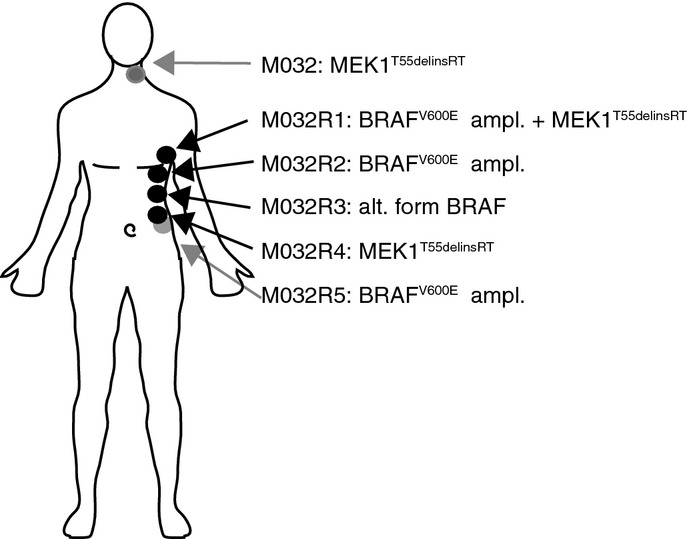
Treatment schedule of patient M032, who received a PET scan two weeks before the start of the treatment (purple asterisk), followed by a baseline CT scan and every two months a follow-up CT scan (red asterisks). Metastases were surgically removed either shortly before the start of the treatment or after resistance occurred (post, M032R1-R5). PR, partial response; PD, progressive disease. Locations of the removed metastases are indicated in the illustration.
Examples of CT scan images of several metastases (M032R1, M032R2, and M032R5).
Volumetric measurements based on the PET and CT scans could be generated from M032, M032R1, and M032R5. Graph illustrates that the metastases R1 and R5 initially expanded before the start of the treatment, but regressed upon vemurafenib treatment. M032R5 showed progressive disease after 4 months, whereas M032R1 still displayed stable disease. M032 was excised and no recurrence was observed.
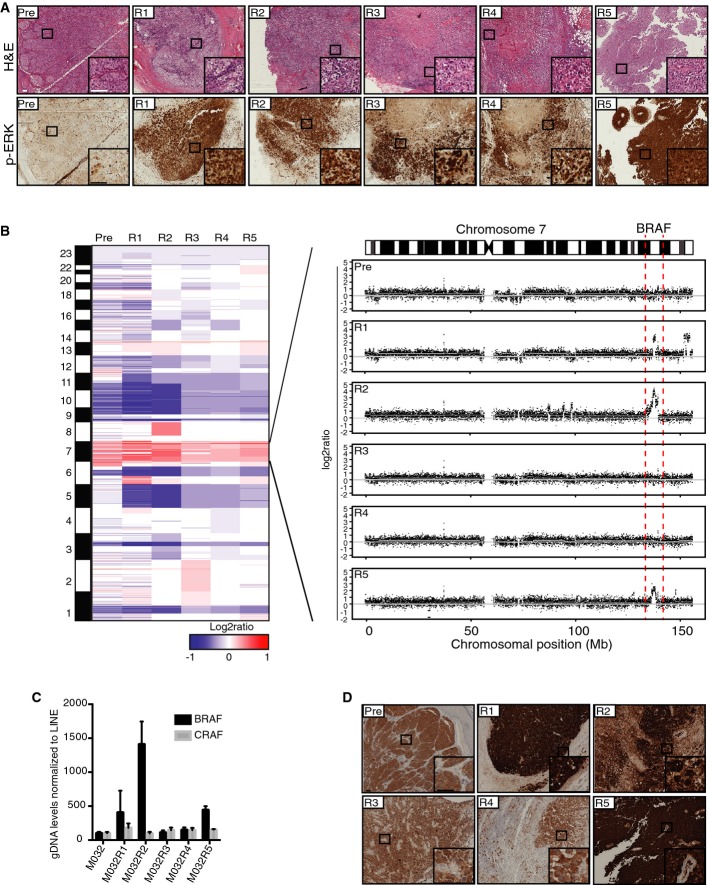
Hematoxylin–eosin (H&E) and p-ERK stainings on FFPE material of all metastases showed that all vemurafenib-resistant tumors had reactivation of the MAPK pathway. Scale bar represents 100 μm.
CNA profiles were generated from the WES data with germ line DNA as a reference. Colors represent segmented log2 ratio values with red for gain and blue for loss. Further inspection of chromosome 7 where BRAF is located revealed amplification (7q34) of this region in three vemurafenib-resistant metastases, namely M032R1, M032R2, and M032R5.
qPCR was performed on gDNA retrieved from each of the metastases, using primers for BRAF and CRAF and normalized on LINE levels. Bars represent the mean of three replicates, error bars indicate standard deviation. The results confirmed that BRAF was amplified in M032R1, M032R2, and M032R5.
Staining for BRAFV600E with a mutant epitope-specific antibody confirmed the upregulation of BRAFV600E in R1, R2, and R5. Scale bar represents 100 μm.
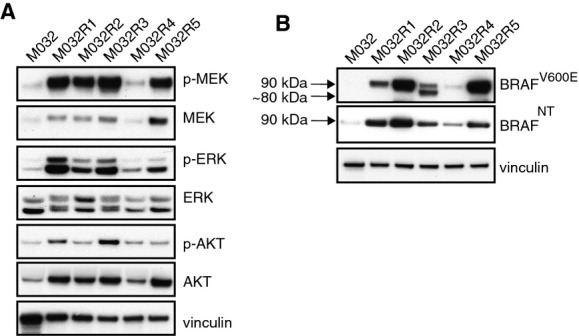
Immunoblotting for MAPK pathway components confirmed reactivation of p-ERK and p-MEK in all resistant metastases.
Immunoblotting for M032 patient’s samples using an antibody directed against either the mutant BRAFV600E epitope or an N-terminal region of BRAF revealed the expression of a shorter variant of BRAFV600E in M032R3, of approximately 80 kDa. This 80-kDa band could not be detected with the N-terminus-recognizing antibody.
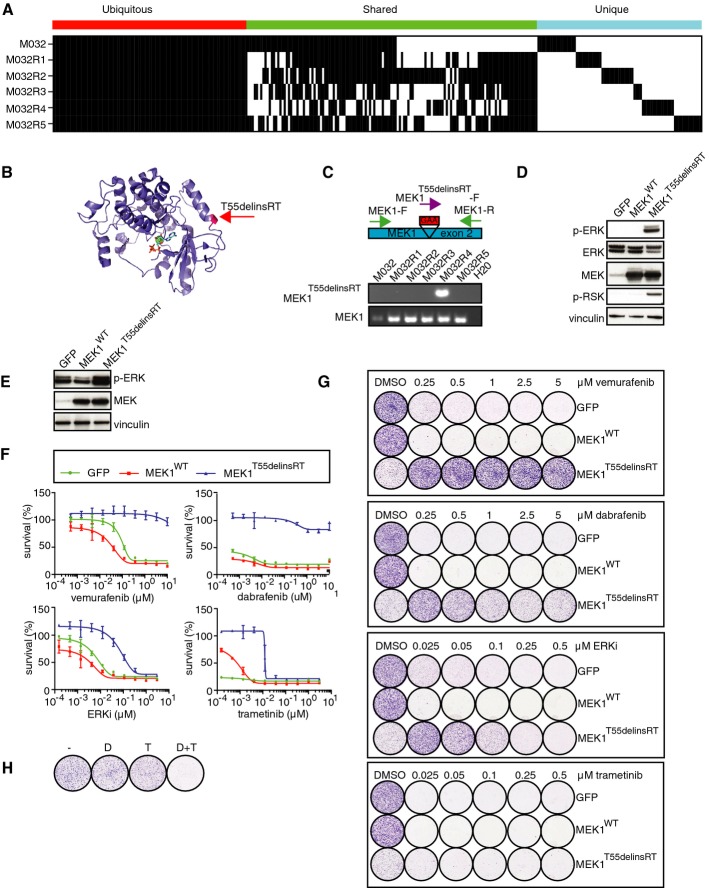
Somatic mutations present in each of these metastases, revealing that mutations were either shared by the pre-treatment tumor and all metastases, by some metastases, or only by single metastases.
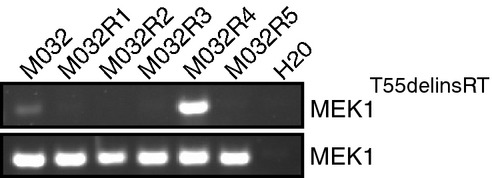
A 3-bp in-frame insertion in MEK1 (T55delinsRT) was identified in M032R4. This mutation was located at the base of the α-helix at the N-terminal site of MEK1.
Validation of T55delinsRT was done by PCR using insertion-specific primers. As a control, primers amplifying exon 2 of MEK1WT were used.
Transfection of pQXCIP-GFP, MEK1-WT, or MEK1-T55delinsRT into HEK293T cells showed that this mutation induces p-ERK and p-RSK.
Validation of expression of pQXCIP-GFP, MEK1WT, or MEK1T55delinsRT in A375 melanoma cells by immunoblotting.
Dose–response curves for A375 melanoma cells expressing GFP, MEK1WT, or MEK1T55delinsRT with indicated doses of BRAF inhibitor vemurafenib, ERK inhibitor SCH772984, or MEK inhibitor trametinib. Error bars indicate standard deviation.
Colony formation assays with A375 melanoma cells, infected with GFP, MEK1WT, or MEK1T55delinsRT-encoding lentivirus, and treated with indicated doses and inhibitors.
Treatment of A375 MEK1T55delinsRT melanoma cells with DMSO (−), 250 nM dabrafenib (D), 10 nM trametinib (T), or a combination (D+T).
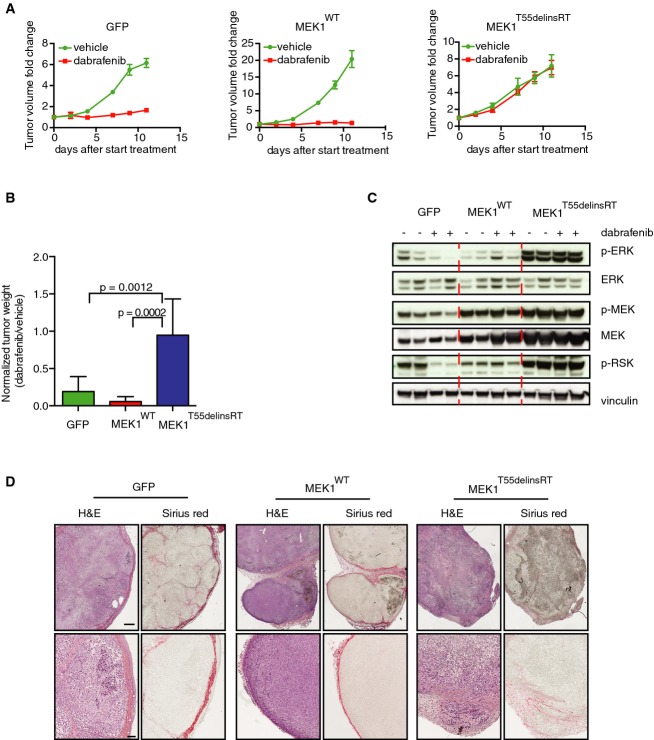
A375 melanoma cells expressing GFP, MEK1WT, or MEK1T55delinsRT were injected into immune-deficient mice (n = 8 per group), and after the tumor size of ˜100 mm3 was reached, mice were treated with 30 mg/kg dabrafenib or vehicle. Graphs represent fold change in tumor volume normalized on the tumor volume on the day of the start of the treatment. Error bars indicate standard error of the mean.
Average tumor weight of all groups at the end of the experiment described in (A). Error bars indicate standard deviation. Unpaired t-test was used to calculate statistical significance. P-values are indicated in the graph.
Immunoblotting for MAPK pathway components performed on tumors of all groups.
Immunohistochemistry for Sirius Red (staining for collagen) and H&E on tumors from all groups, to identify grade of demarcation and local invasion. Scale bar represents 500 (upper row) and 100 (lower row) μm.
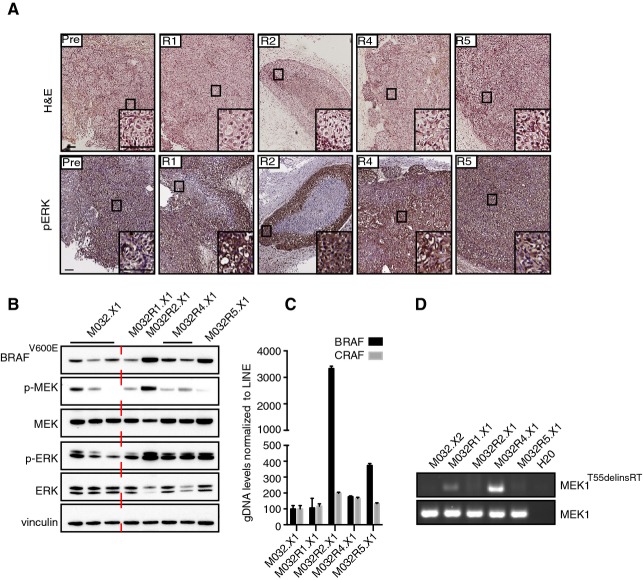
Staining for hematoxylin–eosin (H&E) and p-ERK on the PDX samples M032.X1, M032R1.X1, M032R2.X1, M032R4.X1, and M032R5.X1. Stainings showed that p-ERK is higher in the PDX derived from the resistant metastases. Scale bar represents 100 μm.
Immunoblotting for components of the MAPK pathway confirmed reactivation of p-ERK in the PDX derived from the vemurafenib-resistant metastases, although the p-ERK signal is heterogeneous in the pre-treatment PDX.
Validation of the BRAF amplification was performed by qPCR on gDNA derived from the PDX, using primers for BRAF and CRAF and normalized on LINE expression. Bars represent the mean of three replicates, error bars indicate standard deviation. The results confirmed that BRAF was amplified in PDX derived from M032R2 and M032R5, but not from M032R1.
Presence of the MEK1T55delinsRT was analyzed by PCR using insertion-specific primers. As a control, primers amplifying exon 2 of MEK1WT were used. The insertion was found in M032R1.X1 and M032R4.X1.
Comment in
-
Spatial intra-tumour heterogeneity in acquired resistance to targeted therapy complicates the use of PDX models for co-clinical cancer studies.EMBO Mol Med. 2015 Sep;7(9):1087-9. doi: 10.15252/emmm.201505431. EMBO Mol Med. 2015. PMID: 26174485 Free PMC article.
Similar articles
-
Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression.Clin Cancer Res. 2013 Oct 15;19(20):5749-57. doi: 10.1158/1078-0432.CCR-13-0661. Epub 2013 Aug 15. Clin Cancer Res. 2013. PMID: 23948972
-
The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma.Expert Opin Drug Discov. 2016 Sep;11(9):907-16. doi: 10.1080/17460441.2016.1201057. Epub 2016 Jun 23. Expert Opin Drug Discov. 2016. PMID: 27327499 Free PMC article. Review.
-
Vemurafenib in patients with BRAF V600E mutation-positive advanced melanoma.Clin Ther. 2012 Jul;34(7):1474-86. doi: 10.1016/j.clinthera.2012.06.009. Epub 2012 Jun 27. Clin Ther. 2012. PMID: 22742884 Review.
-
Differential inhibition of ex-vivo tumor kinase activity by vemurafenib in BRAF(V600E) and BRAF wild-type metastatic malignant melanoma.PLoS One. 2013 Aug 30;8(8):e72692. doi: 10.1371/journal.pone.0072692. eCollection 2013. PLoS One. 2013. PMID: 24023633 Free PMC article.
-
Spatial intra-tumour heterogeneity in acquired resistance to targeted therapy complicates the use of PDX models for co-clinical cancer studies.EMBO Mol Med. 2015 Sep;7(9):1087-9. doi: 10.15252/emmm.201505431. EMBO Mol Med. 2015. PMID: 26174485 Free PMC article.
Cited by
-
Implementation of a Multicenter Biobanking Collaboration for Next-Generation Sequencing-Based Biomarker Discovery Based on Fresh Frozen Pretreatment Tumor Tissue Biopsies.Oncologist. 2017 Jan;22(1):33-40. doi: 10.1634/theoncologist.2016-0085. Epub 2016 Sep 23. Oncologist. 2017. PMID: 27662884 Free PMC article. Clinical Trial.
-
Modeling neoplastic disease with spheroids and organoids.J Hematol Oncol. 2020 Jul 16;13(1):97. doi: 10.1186/s13045-020-00931-0. J Hematol Oncol. 2020. PMID: 32677979 Free PMC article. Review.
-
Xenograft and organoid model systems in cancer research.EMBO J. 2019 Aug 1;38(15):e101654. doi: 10.15252/embj.2019101654. Epub 2019 Jul 8. EMBO J. 2019. PMID: 31282586 Free PMC article. Review.
-
Autophagy Inhibition in BRAF-Driven Cancers.Cancers (Basel). 2021 Jul 13;13(14):3498. doi: 10.3390/cancers13143498. Cancers (Basel). 2021. PMID: 34298710 Free PMC article. Review.
-
Tumour Cell Heterogeneity.F1000Res. 2016 Feb 29;5:F1000 Faculty Rev-238. doi: 10.12688/f1000research.7210.1. eCollection 2016. F1000Res. 2016. PMID: 26973786 Free PMC article. Review.
References
-
- Bertolotto C, Lesueur F, Giuliano S, Strub T, deLichy M, Bille K, Dessen P, d’Hayer B, Mohamdi H, Remenieras A, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480:94–98. - PubMed
-
- Carlino MS, Fung C, Shahheydari H, Todd JR, Boyd SC, Irvine M, Nagrial AM, Scolyer RA, Kefford RF, Long GV, et al. Preexisting MEK1P124 mutations diminish response to BRAF inhibitors in metastatic melanoma patients. Clin Cancer Res. 2015;21:98–105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
