

Dissection of a Complex Disease Susceptibility Region Using a Bayesian Stochastic Search Approach to Fine Mapping
- PMID: 26106896
- PMCID: PMC4481316
- DOI: 10.1371/journal.pgen.1005272
Dissection of a Complex Disease Susceptibility Region Using a Bayesian Stochastic Search Approach to Fine Mapping
Abstract
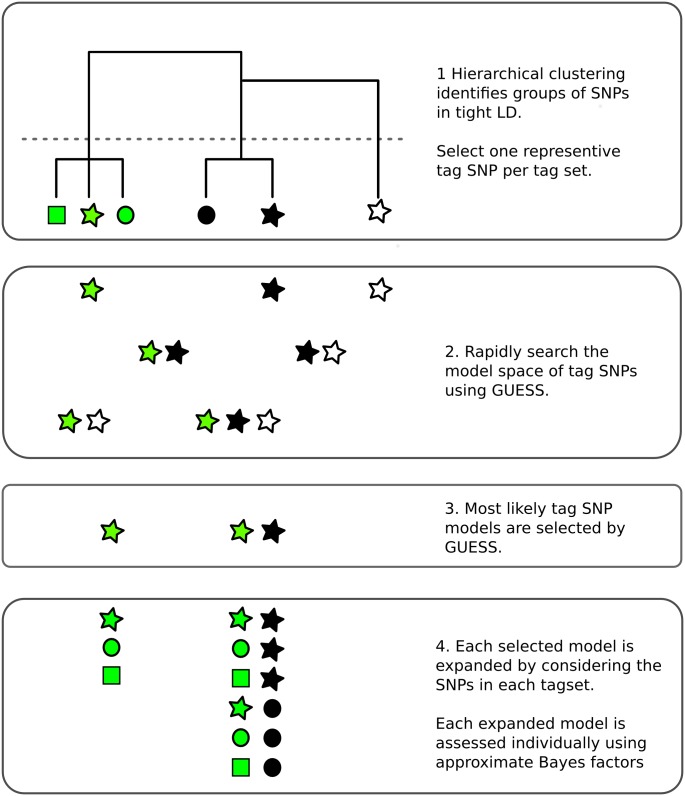
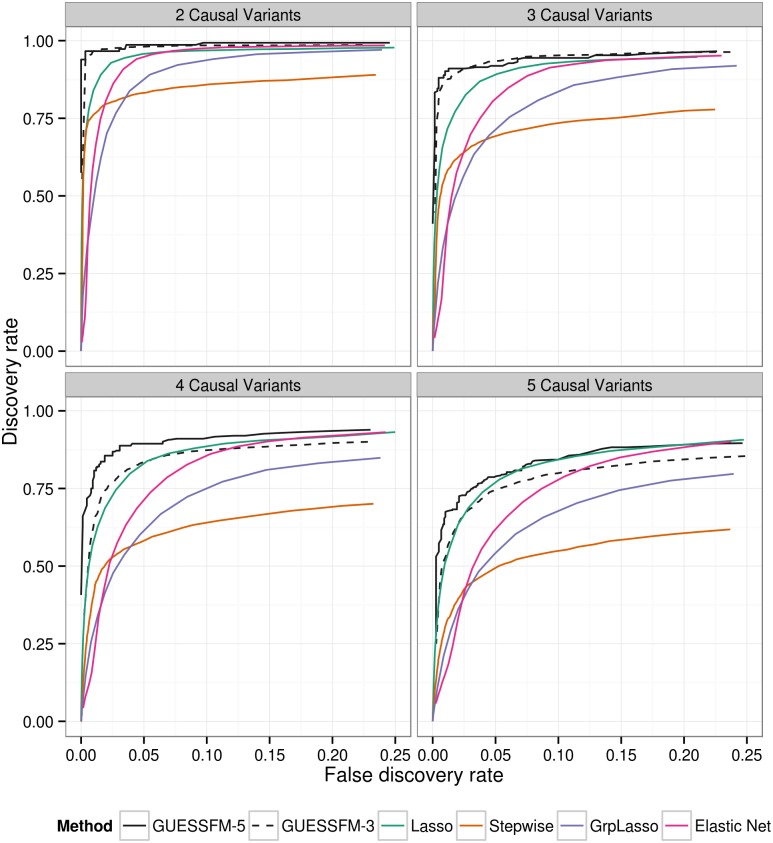
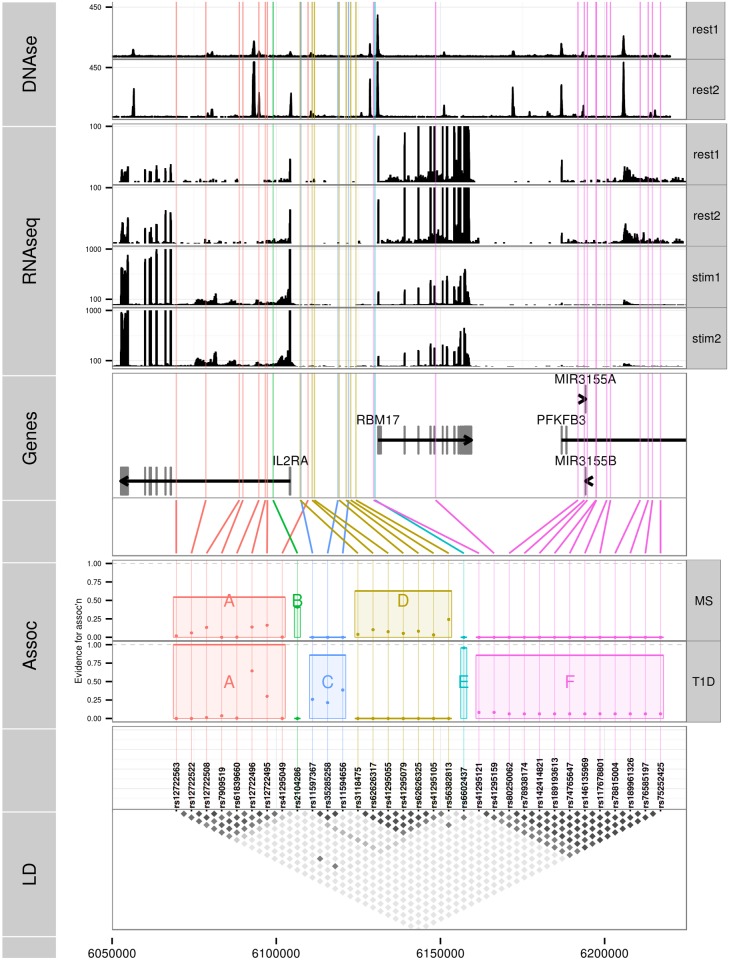
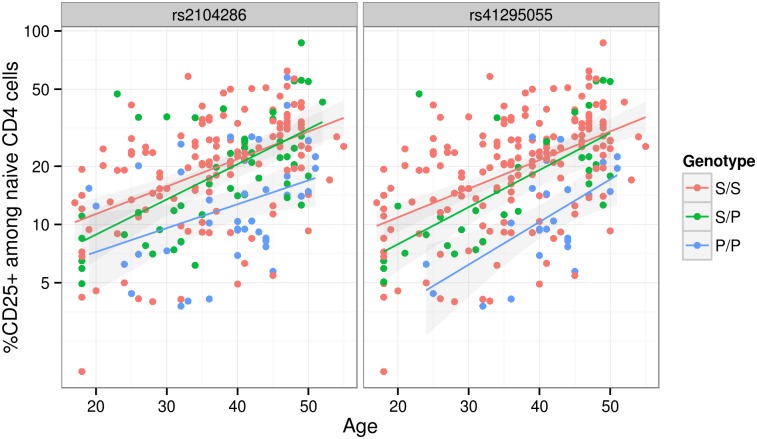
Identification of candidate causal variants in regions associated with risk of common diseases is complicated by linkage disequilibrium (LD) and multiple association signals. Nonetheless, accurate maps of these variants are needed, both to fully exploit detailed cell specific chromatin annotation data to highlight disease causal mechanisms and cells, and for design of the functional studies that will ultimately be required to confirm causal mechanisms. We adapted a Bayesian evolutionary stochastic search algorithm to the fine mapping problem, and demonstrated its improved performance over conventional stepwise and regularised regression through simulation studies. We then applied it to fine map the established multiple sclerosis (MS) and type 1 diabetes (T1D) associations in the IL-2RA (CD25) gene region. For T1D, both stepwise and stochastic search approaches identified four T1D association signals, with the major effect tagged by the single nucleotide polymorphism, rs12722496. In contrast, for MS, the stochastic search found two distinct competing models: a single candidate causal variant, tagged by rs2104286 and reported previously using stepwise analysis; and a more complex model with two association signals, one of which was tagged by the major T1D associated rs12722496 and the other by rs56382813. There is low to moderate LD between rs2104286 and both rs12722496 and rs56382813 (r2 ≃ 0:3) and our two SNP model could not be recovered through a forward stepwise search after conditioning on rs2104286. Both signals in the two variant model for MS affect CD25 expression on distinct subpopulations of CD4+ T cells, which are key cells in the autoimmune process. The results support a shared causal variant for T1D and MS. Our study illustrates the benefit of using a purposely designed model search strategy for fine mapping and the advantage of combining disease and protein expression data.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Miller AJ (1984) Selection of subsets of regression variables. Journal of the Royal Statistical Society Series A (General) 147: pp. 389–425. 10.2307/2981576 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
