

A roadmap to multifactor dimensionality reduction methods
- PMID: 26108231
- PMCID: PMC4793893
- DOI: 10.1093/bib/bbv038
A roadmap to multifactor dimensionality reduction methods
Abstract
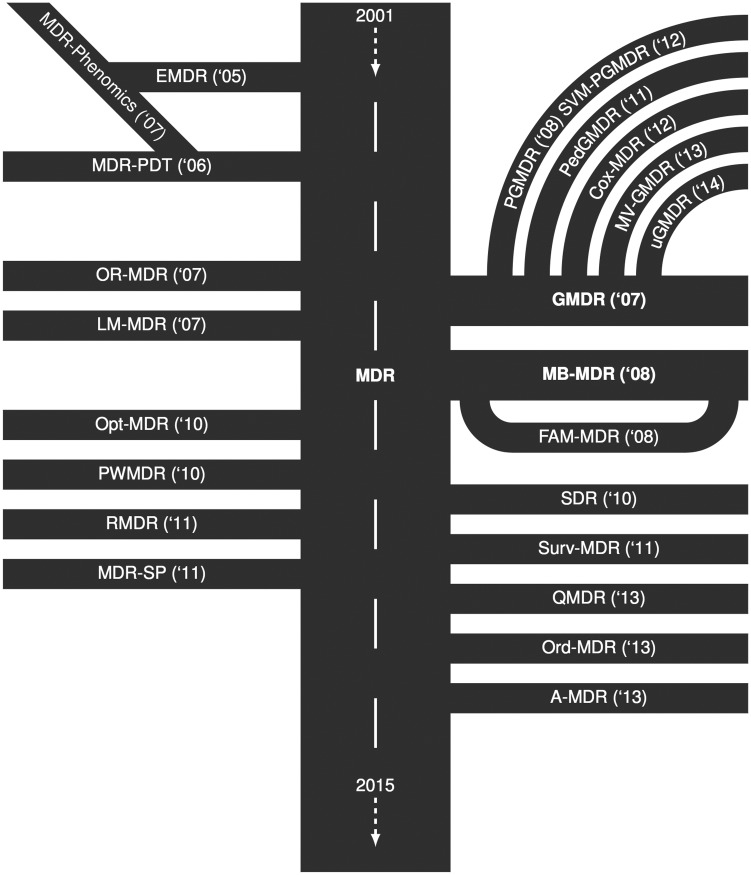
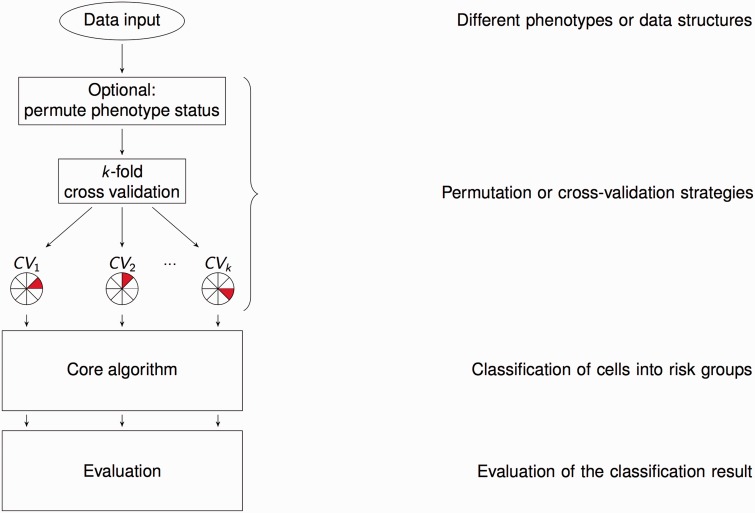
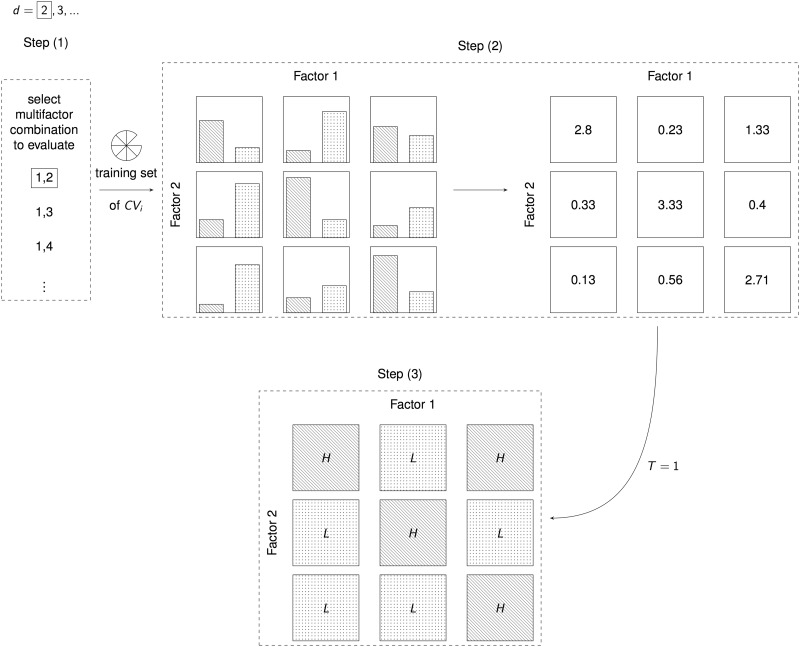
Complex diseases are defined to be determined by multiple genetic and environmental factors alone as well as in interactions. To analyze interactions in genetic data, many statistical methods have been suggested, with most of them relying on statistical regression models. Given the known limitations of classical methods, approaches from the machine-learning community have also become attractive. From this latter family, a fast-growing collection of methods emerged that are based on the Multifactor Dimensionality Reduction (MDR) approach. Since its first introduction, MDR has enjoyed great popularity in applications and has been extended and modified multiple times. Based on a literature search, we here provide a systematic and comprehensive overview of these suggested methods. The methods are described in detail, and the availability of implementations is listed. Most recent approaches offer to deal with large-scale data sets and rare variants, which is why we expect these methods to even gain in popularity.
Keywords: data mining; epistasis; interaction; machine learning; multifactor dimensionality reduction.
© The Author 2015. Published by Oxford University Press.
Figures
References
-
- Cho YM, Ritchie MD, Moore JH, et al. Multifactor-dimensionality reduction shows a two-locus interaction associated with Type 2 diabetes mellitus. Diabetologia 2004;47:549–54. - PubMed
-
- Tsai CT, Lai LP, Lin JL, et al. Renin-angiotensin system gene polymorphisms and atrial fibrillation. Circulation 2004;109:1640–6. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
