

Comparison of RNA-seq and microarray-based models for clinical endpoint prediction
- PMID: 26109056
- PMCID: PMC4506430
- DOI: 10.1186/s13059-015-0694-1
Comparison of RNA-seq and microarray-based models for clinical endpoint prediction
Abstract
Background: Gene expression profiling is being widely applied in cancer research to identify biomarkers for clinical endpoint prediction. Since RNA-seq provides a powerful tool for transcriptome-based applications beyond the limitations of microarrays, we sought to systematically evaluate the performance of RNA-seq-based and microarray-based classifiers in this MAQC-III/SEQC study for clinical endpoint prediction using neuroblastoma as a model.
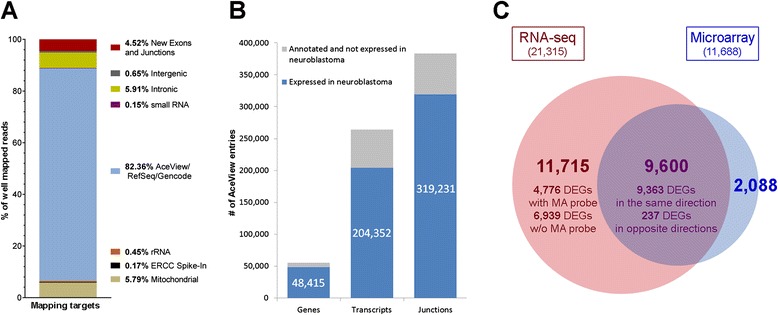
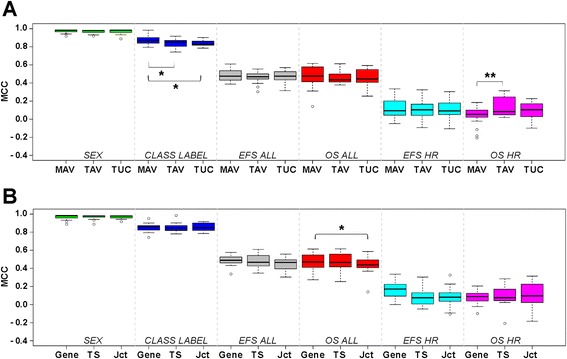
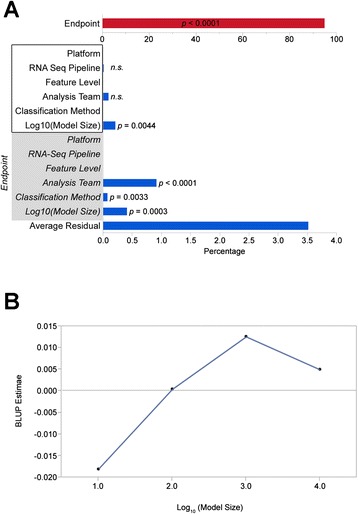
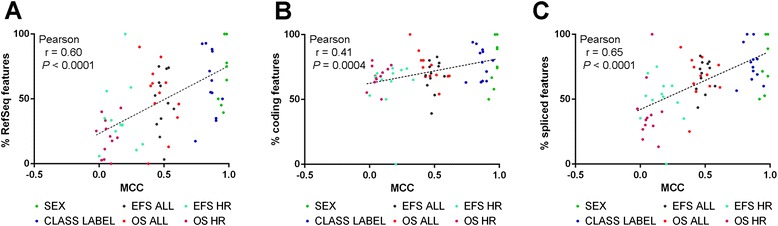
Results: We generate gene expression profiles from 498 primary neuroblastomas using both RNA-seq and 44 k microarrays. Characterization of the neuroblastoma transcriptome by RNA-seq reveals that more than 48,000 genes and 200,000 transcripts are being expressed in this malignancy. We also find that RNA-seq provides much more detailed information on specific transcript expression patterns in clinico-genetic neuroblastoma subgroups than microarrays. To systematically compare the power of RNA-seq and microarray-based models in predicting clinical endpoints, we divide the cohort randomly into training and validation sets and develop 360 predictive models on six clinical endpoints of varying predictability. Evaluation of factors potentially affecting model performances reveals that prediction accuracies are most strongly influenced by the nature of the clinical endpoint, whereas technological platforms (RNA-seq vs. microarrays), RNA-seq data analysis pipelines, and feature levels (gene vs. transcript vs. exon-junction level) do not significantly affect performances of the models.
Conclusions: We demonstrate that RNA-seq outperforms microarrays in determining the transcriptomic characteristics of cancer, while RNA-seq and microarray-based models perform similarly in clinical endpoint prediction. Our findings may be valuable to guide future studies on the development of gene expression-based predictive models and their implementation in clinical practice.
Figures

References
-
- Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–24. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
