

Comparison of GENCODE and RefSeq gene annotation and the impact of reference geneset on variant effect prediction
- PMID: 26110515
- PMCID: PMC4502323
- DOI: 10.1186/1471-2164-16-S8-S2
Comparison of GENCODE and RefSeq gene annotation and the impact of reference geneset on variant effect prediction
Abstract
Background: A vast amount of DNA variation is being identified by increasingly large-scale exome and genome sequencing projects. To be useful, variants require accurate functional annotation and a wide range of tools are available to this end. McCarthy et al recently demonstrated the large differences in prediction of loss-of-function (LoF) variation when RefSeq and Ensembl transcripts are used for annotation, highlighting the importance of the reference transcripts on which variant functional annotation is based.
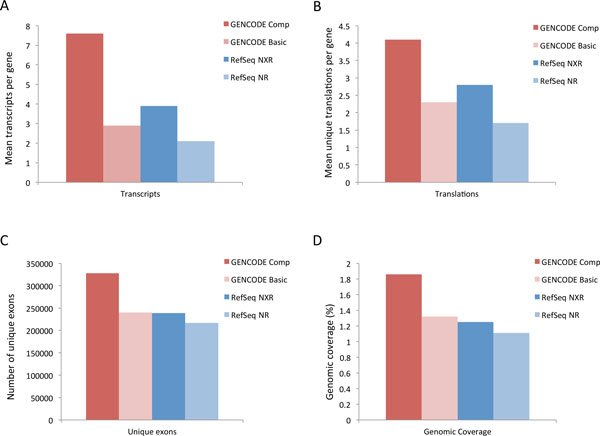
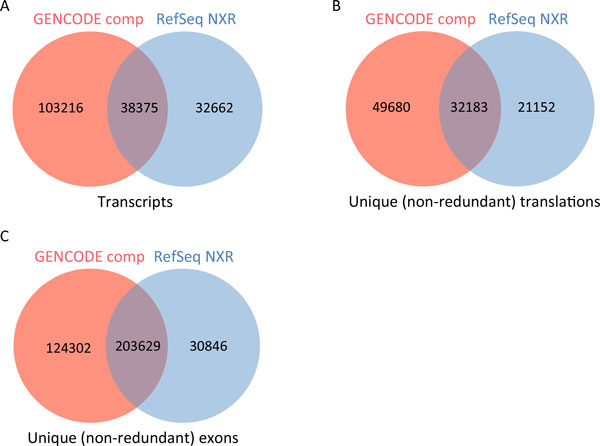
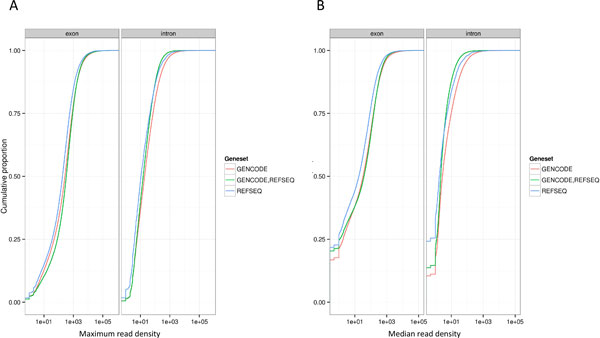
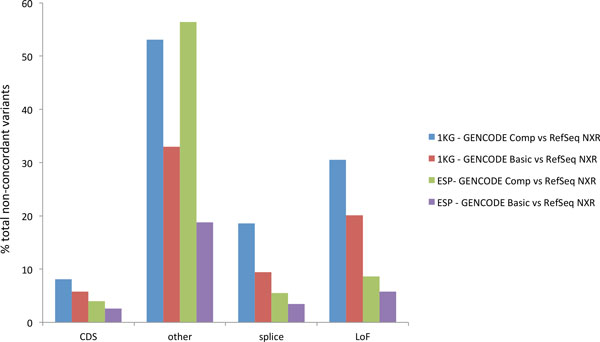
Results: We describe a detailed analysis of the similarities and differences between the gene and transcript annotation in the GENCODE and RefSeq genesets. We demonstrate that the GENCODE Comprehensive set is richer in alternative splicing, novel CDSs, novel exons and has higher genomic coverage than RefSeq, while the GENCODE Basic set is very similar to RefSeq. Using RNAseq data we show that exons and introns unique to one geneset are expressed at a similar level to those common to both. We present evidence that the differences in gene annotation lead to large differences in variant annotation where GENCODE and RefSeq are used as reference transcripts, although this is predominantly confined to non-coding transcripts and UTR sequence, with at most ~30% of LoF variants annotated discordantly. We also describe an investigation of dominant transcript expression, showing that it both supports the utility of the GENCODE Basic set in providing a smaller set of more highly expressed transcripts and provides a useful, biologically-relevant filter for further reducing the complexity of the transcriptome.
Conclusions: The reference transcripts selected for variant functional annotation do have a large effect on the outcome. The GENCODE Comprehensive transcripts contain more exons, have greater genomic coverage and capture many more variants than RefSeq in both genome and exome datasets, while the GENCODE Basic set shows a higher degree of concordance with RefSeq and has fewer unique features. We propose that the GENCODE Comprehensive set has great utility for the discovery of new variants with functional potential, while the GENCODE Basic set is more suitable for applications demanding less complex interpretation of functional variants.
Figures
References
-
- UK10K: Rare Genetic Variants in Health and Disease (2010-2013) http://www.uk10k.org http://www.uk10k.org
-
- Futema M, Plagnol V, Li K, Whittall RA, Neil HA, Seed M, Simon Broome C, Bertolini S, Calandra S, Descamps OS. et al.Whole exome sequencing of familial hypercholesterolaemia patients negative for LDLR/APOB/PCSK9 mutations. J Med Genet. 2014;51(8):537–544. doi: 10.1136/jmedgenet-2014-102405. - DOI - PMC - PubMed
-
- 100,000 Genomes Project. http://www.genomicsengland.co.uk http://www.genomicsengland.co.uk
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
