

Application of single-cell genomics in cancer: promise and challenges
- PMID: 26113645
- PMCID: PMC4571998
- DOI: 10.1093/hmg/ddv235
Application of single-cell genomics in cancer: promise and challenges
Abstract
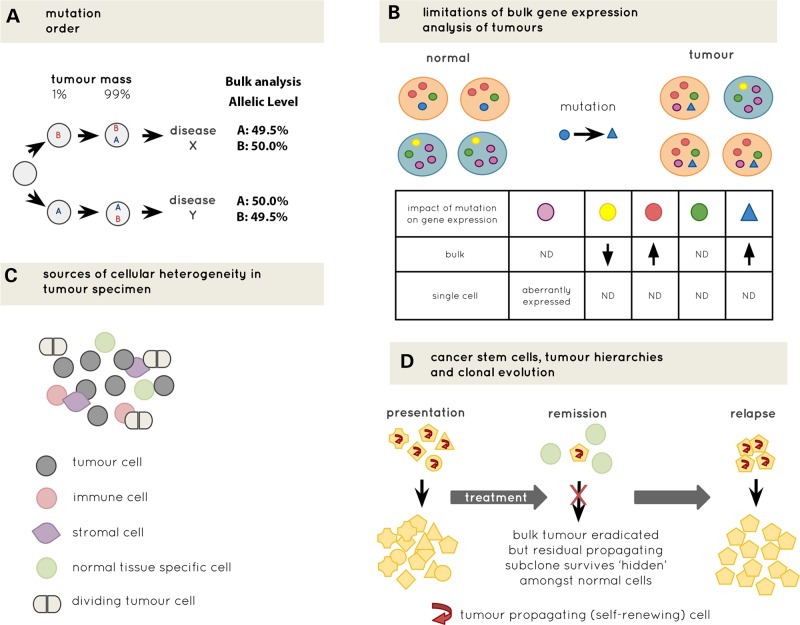
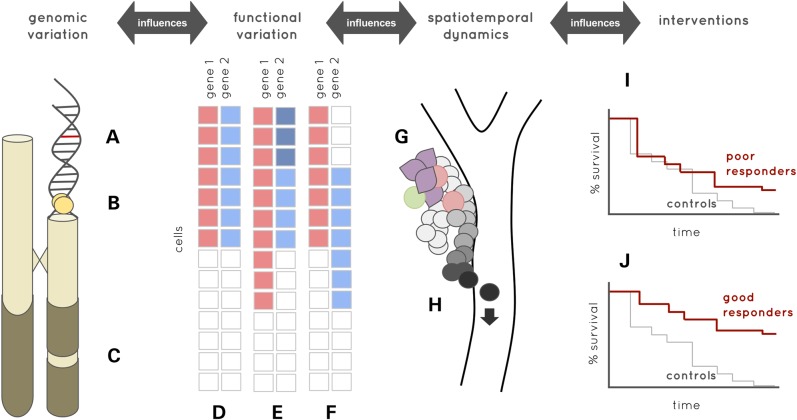
Recent advances in single-cell genomics are opening up unprecedented opportunities to transform cancer genomics. While bulk tissue genomic analysis across large populations of tumour cells has provided key insights into cancer biology, this approach does not provide the resolution that is critical for understanding the interaction between different genetic events within the cellular hierarchy of the tumour during disease initiation, evolution, relapse and metastasis. Single-cell genomic approaches are uniquely placed to definitively unravel complex clonal structures and tissue hierarchies, account for spatiotemporal cell interactions and discover rare cells that drive metastatic disease, drug resistance and disease progression. Here we present five challenges that need to be met for single-cell genomics to fulfil its potential as a routine tool alongside bulk sequencing. These might be thought of as being challenges related to samples (processing and scale for analysis), sensitivity and specificity of mutation detection, sources of heterogeneity (biological and technical), synergies (from data integration) and systems modelling. We discuss these in the context of recent advances in technologies and data modelling, concluding with implications for moving cancer research into the clinic.
© The Author 2015. Published by Oxford University Press.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
