

Integration of Downstream Signals of Insulin-like Growth Factor-1 Receptor by Endoplasmic Reticulum Stress for Estrogen-Induced Growth or Apoptosis in Breast Cancer Cells
- PMID: 26116171
- PMCID: PMC4763612
- DOI: 10.1158/1541-7786.MCR-14-0494
Integration of Downstream Signals of Insulin-like Growth Factor-1 Receptor by Endoplasmic Reticulum Stress for Estrogen-Induced Growth or Apoptosis in Breast Cancer Cells
Abstract
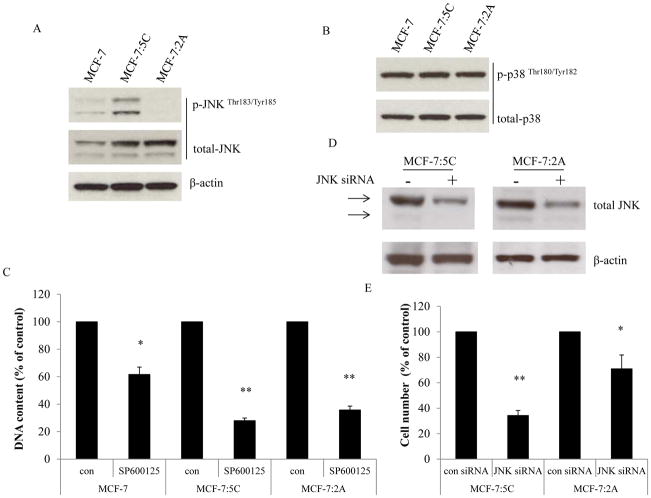
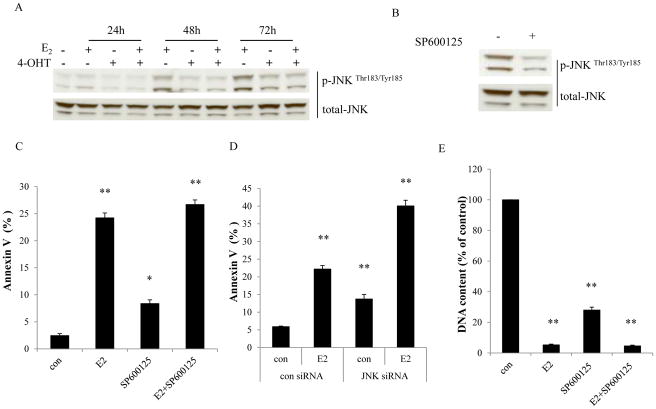
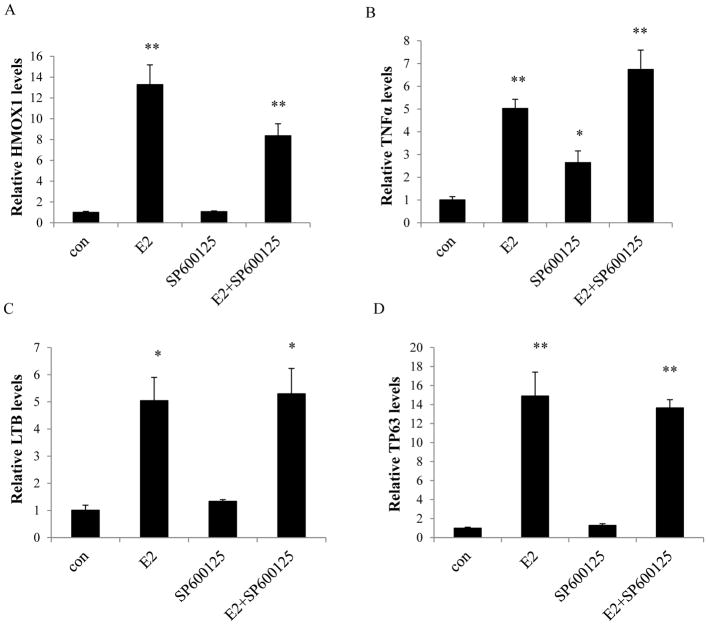
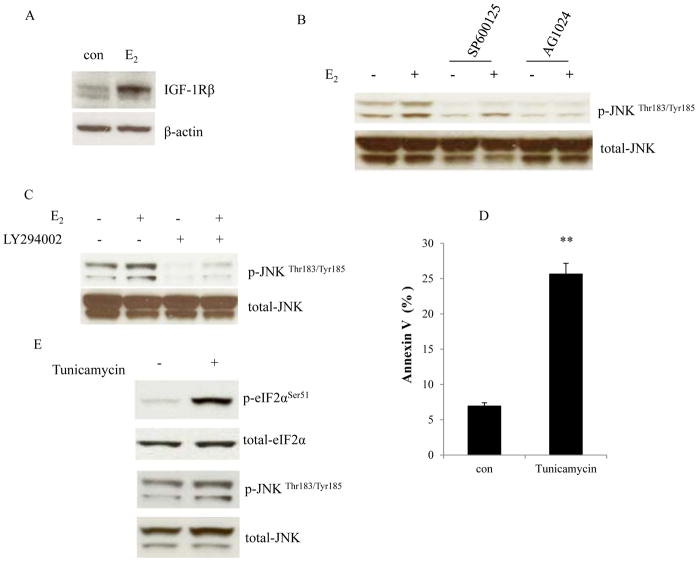
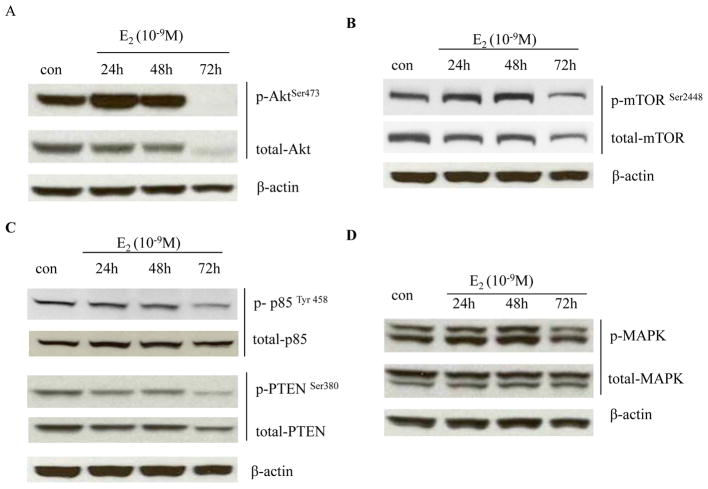
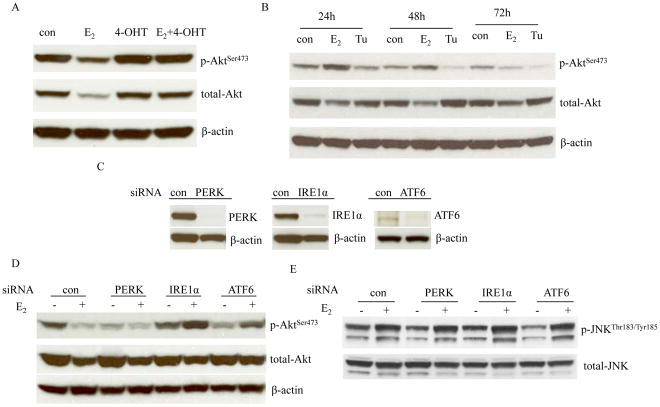
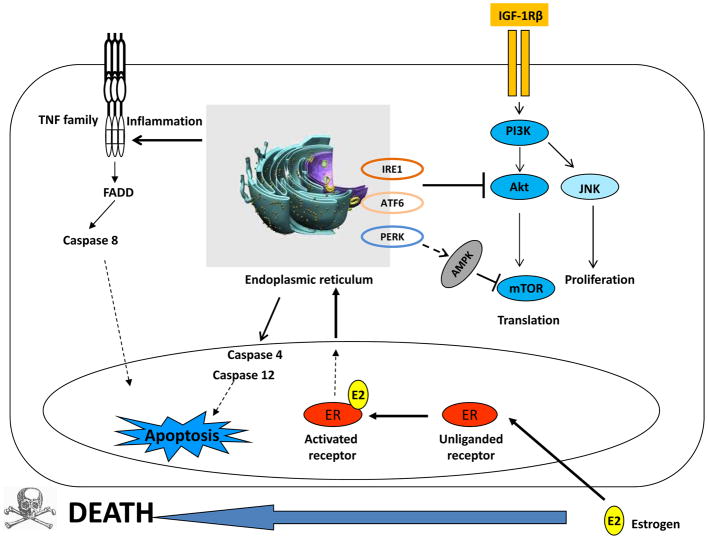
Estrogen (E2) exerts a dual function on E2-deprived breast cancer cells, with both initial proliferation and subsequent induction of stress responses to cause apoptosis. However, the mechanism by which E2 integrally regulates cell growth or apoptosis-associated pathways remains to be elucidated. Here, E2 deprivation results in many alterations in stress-responsive pathways. For instance, E2-deprived breast cancer cells had higher basal levels of stress-activated protein kinase, c-Jun N-terminal kinase (JNK), compared with wild-type MCF-7 cells. E2 treatment further constitutively activated JNK after 24 hours. However, inhibition of JNK (SP600125) was unable to abolish E2- induced apoptosis, whereas SP600125 alone arrested cells at the G2 phase of the cell cycle and increased apoptosis. Further examination showed that inhibition of JNK increased gene expression of TNFα and did not effectively attenuate expression of apoptosis-related genes induced by E2. A notable finding was that E2 regulated both JNK and Akt as the downstream signals of insulin-like growth factor-1 receptor (IGFIR)/PI3K, but with distinctive modulation patterns: JNK was constitutively activated, whereas Akt and Akt-associated proteins, such as PTEN and mTOR, were selectively degraded. Endoplasmic reticulum-associated degradation (ERAD) was involved in the selective protein degradation. These findings highlight a novel IGFIR/PI3K/JNK axis that plays a proliferative role during the prelude to E2-induced apoptosis and that the endoplasmic reticulum is a key regulatory site to decide cell fate after E2 treatment.
Implications: This study provides a new rationale for further exploration of E2-induced apoptosis to improve clinical benefit.
©2015 American Association for Cancer Research.
Conflict of interest statement
There are no conflicts to disclose.
Figures
References
-
- Wolf DM, Jordan VC. A laboratory model to explain the survival advantage observed in patients taking adjuvant tamoxifen therapy. Recent Results Cancer Res. 1993;127:23–33. - PubMed
-
- Yao K, Lee ES, Bentrem DJ, England G, Schafer JI, O’Regan RM, et al. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res. 2000;6:2028–36. - PubMed
-
- Song RX, Mor G, Naftolin F, McPherson RA, Song J, Zhang Z, et al. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17beta-estradiol. J Natl Cancer Inst. 2001;93:1714–23. - PubMed
-
- Lewis JS, Meeke K, Osipo C, Ross EA, Kidawi N, Li T, et al. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst. 2005;97:1746–59. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
