

Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study
- PMID: 26116186
- PMCID: PMC4579482
- DOI: 10.1016/S1473-3099(15)00062-6
Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study
Erratum in
-
Corrections.Lancet Infect Dis. 2018 Jan;18(1):21. doi: 10.1016/S1473-3099(17)30688-6. Epub 2017 Nov 21. Lancet Infect Dis. 2018. PMID: 29174723 Free PMC article. No abstract available.
Abstract
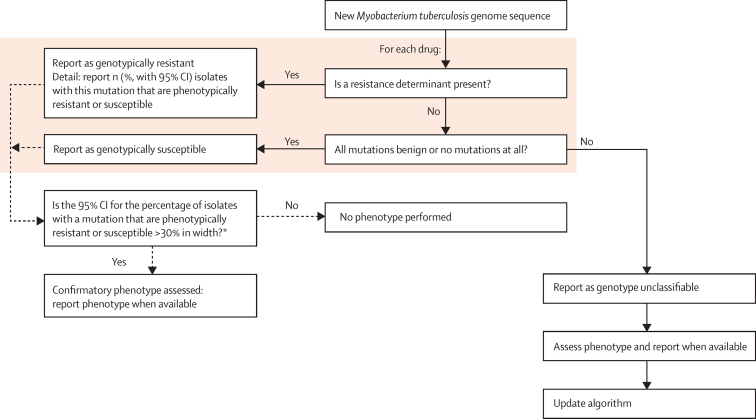
Background: Diagnosing drug-resistance remains an obstacle to the elimination of tuberculosis. Phenotypic drug-susceptibility testing is slow and expensive, and commercial genotypic assays screen only common resistance-determining mutations. We used whole-genome sequencing to characterise common and rare mutations predicting drug resistance, or consistency with susceptibility, for all first-line and second-line drugs for tuberculosis.
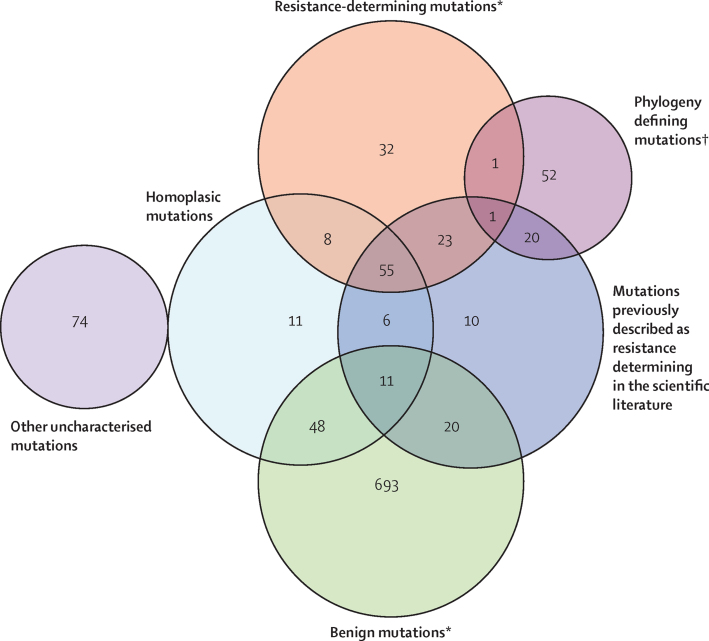
Methods: Between Sept 1, 2010, and Dec 1, 2013, we sequenced a training set of 2099 Mycobacterium tuberculosis genomes. For 23 candidate genes identified from the drug-resistance scientific literature, we algorithmically characterised genetic mutations as not conferring resistance (benign), resistance determinants, or uncharacterised. We then assessed the ability of these characterisations to predict phenotypic drug-susceptibility testing for an independent validation set of 1552 genomes. We sought mutations under similar selection pressure to those characterised as resistance determinants outside candidate genes to account for residual phenotypic resistance.
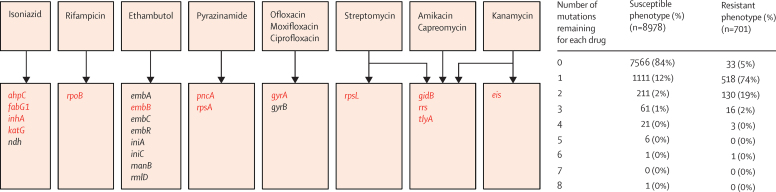
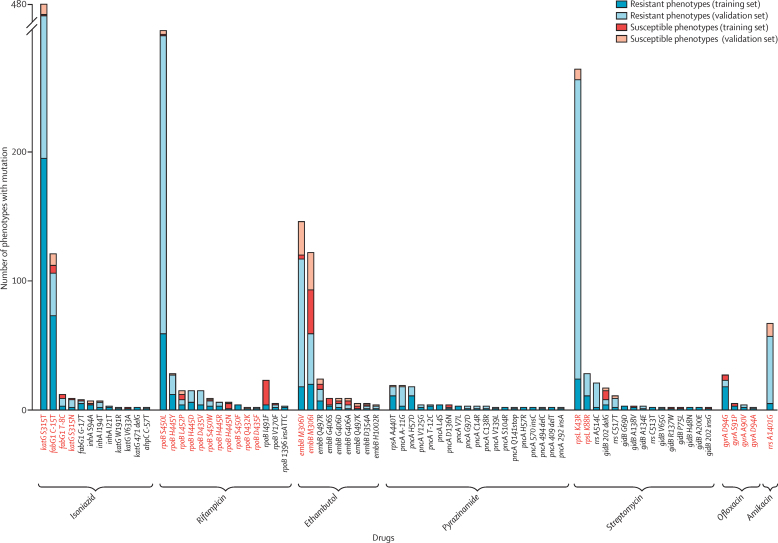
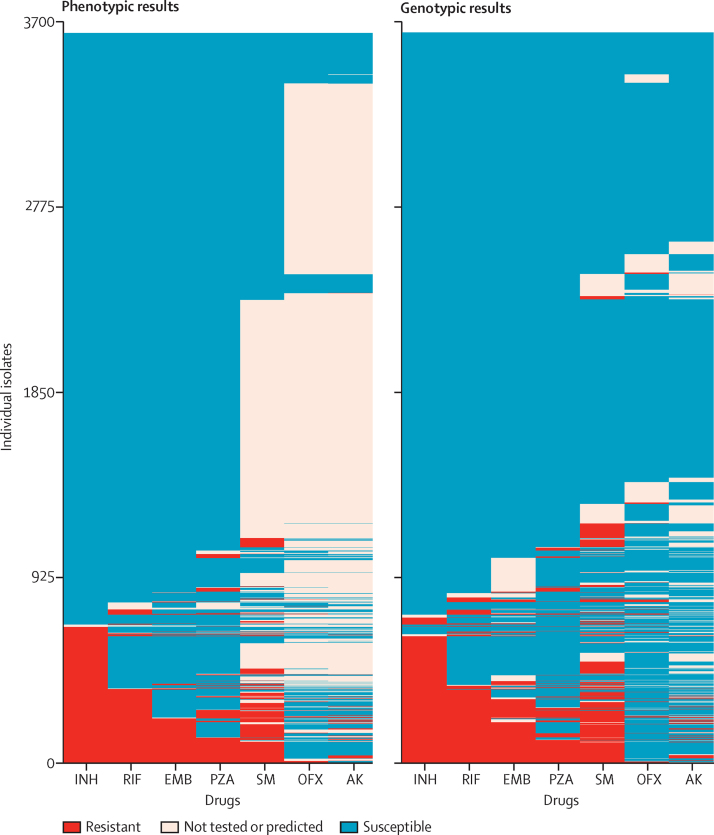
Findings: We characterised 120 training-set mutations as resistance determining, and 772 as benign. With these mutations, we could predict 89·2% of the validation-set phenotypes with a mean 92·3% sensitivity (95% CI 90·7-93·7) and 98·4% specificity (98·1-98·7). 10·8% of validation-set phenotypes could not be predicted because uncharacterised mutations were present. With an in-silico comparison, characterised resistance determinants had higher sensitivity than the mutations from three line-probe assays (85·1% vs 81·6%). No additional resistance determinants were identified among mutations under selection pressure in non-candidate genes.
Interpretation: A broad catalogue of genetic mutations enable data from whole-genome sequencing to be used clinically to predict drug resistance, drug susceptibility, or to identify drug phenotypes that cannot yet be genetically predicted. This approach could be integrated into routine diagnostic workflows, phasing out phenotypic drug-susceptibility testing while reporting drug resistance early.
Funding: Wellcome Trust, National Institute of Health Research, Medical Research Council, and the European Union.
Copyright © 2015 Walker et al. Open Access article distributed under the terms of CC-BY. Published by Elsevier Ltd.. All rights reserved.
Figures
Comment in
-
Towards genomic prediction of drug resistance in tuberculosis.Lancet Infect Dis. 2015 Oct;15(10):1124-1125. doi: 10.1016/S1473-3099(15)00088-2. Epub 2015 Jun 23. Lancet Infect Dis. 2015. PMID: 26116184 No abstract available.
-
Whole-genome sequencing for the diagnosis of drug-resistant tuberculosis.Lancet Infect Dis. 2016 Jan;16(1):17. doi: 10.1016/S1473-3099(15)00474-0. Lancet Infect Dis. 2016. PMID: 26738825 No abstract available.
References
-
- WHO Global tuberculosis report 2014. http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf?... (accessed Nov 28, 2014).
Publication types
MeSH terms
Substances
Grants and funding
- 090532/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- 102541/WT_/Wellcome Trust/United Kingdom
- G0800778/MRC_/Medical Research Council/United Kingdom
- T5-358/WT_/Wellcome Trust/United Kingdom
- 087646/Z/08/Z/WT_/Wellcome Trust/United Kingdom
- DRF-2010-03-40/DH_/Department of Health/United Kingdom
- 087646/WT_/Wellcome Trust/United Kingdom
- 101237/WT_/Wellcome Trust/United Kingdom
- MR/J011398/1/MRC_/Medical Research Council/United Kingdom
- 098051/WT_/Wellcome Trust/United Kingdom
- 098615/WT_/Wellcome Trust/United Kingdom
- IS-HPU-1112-10041/DH_/Department of Health/United Kingdom
- 090532/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
