

EFTUD2 deficiency in vertebrates: Identification of a novel human mutation and generation of a zebrafish model
- PMID: 26118977
- PMCID: PMC4487781
- DOI: 10.1002/bdra.23397
EFTUD2 deficiency in vertebrates: Identification of a novel human mutation and generation of a zebrafish model
Abstract
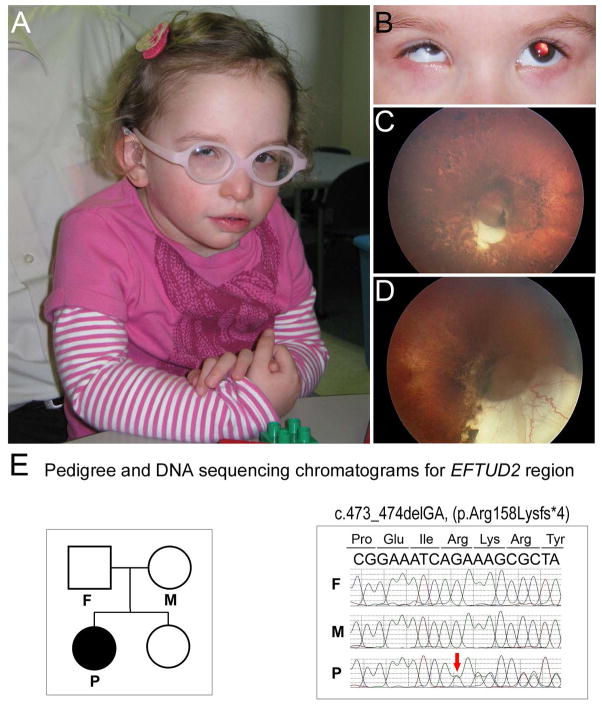
Background: Congenital microphthalmia and coloboma are severe developmental defects that are frequently associated with additional systemic anomalies and display a high level of genetic heterogeneity.
Methods: To identify the pathogenic variant in a patient with microphthalmia, coloboma, retinal dystrophy, microcephaly, and other features, whole exome sequencing analysis of the patient and parental samples was undertaken. To further explore the identified variant/gene, expression and functional studies in zebrafish were performed.
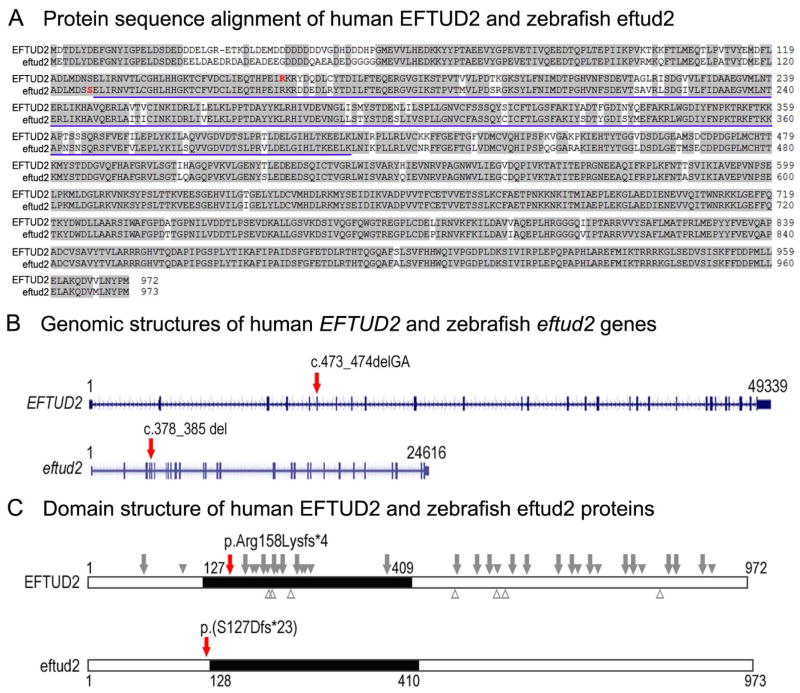
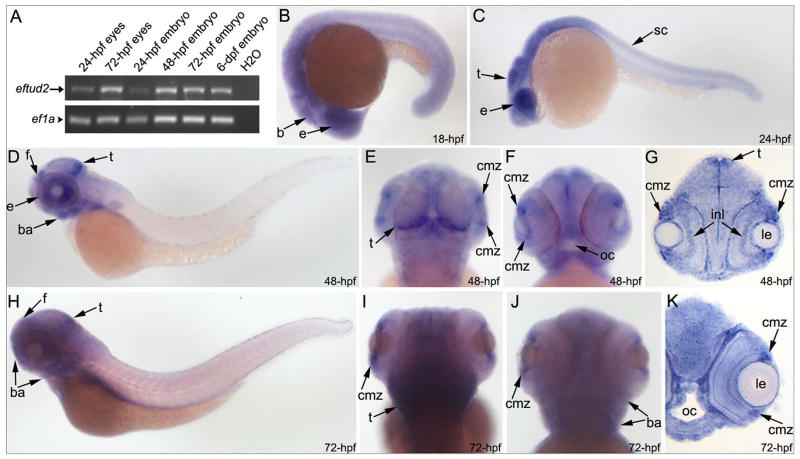
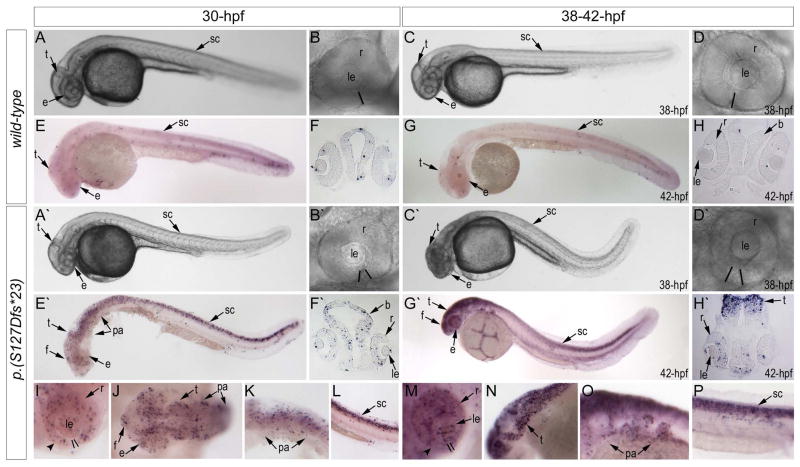
Results: Whole exome sequencing revealed a de novo variant, c.473_474delGA, p.(Arg158Lysfs*4), in EFTUD2 which encodes a component of the spliceosome complex. Dominant mutations in EFTUD2 cause Mandibulofacial Dysostosis, Guion-Almeida type, which does not involve microphthalmia, coloboma, or retinal dystrophy; analysis of genes known to cause these ocular phenotypes identified several variants of unknown significance but no causal alleles in the affected patient. Zebrafish eftud2 demonstrated high sequence conservation with the human gene and broad embryonic expression. TALEN-mediated disruption was employed to generate a c.378_385 del, p.(Ser127Aspfs*23) truncation mutation in eftud2. Homozygous mutants displayed a reduced head size, small eye, curved body, and early embryonic lethality. Apoptosis assays demonstrated a striking increase in terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL)-positive cells in the developing brain, eye, spinal cord, and other tissues starting at 30 hours postfertilization.
Conclusion: This study reports a novel mutation in EFTUD2 in a Mandibulofacial Dysostosis, Guion-Almeida type patient with unusual ocular features and the generation of a first animal model of eftud2 deficiency. The severe embryonic phenotype observed in eftud2 mutants indicates an important conserved role during development of diverse tissues in vertebrates.
Keywords: EFTUD2; coloboma; microphthalmia; retinal dystrophy; zebrafish.
© 2015 Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Bardakjian TM, Schneider A. The genetics of anophthalmia and microphthalmia. Curr Opin Ophthalmol. 2011;22:309–313. - PubMed
-
- Bernier FP, Caluseriu O, Ng S, Schwartzentruber J, Buckingham KJ, Innes AM, Jabs EW, Innis JW, Schuette JL, Gorski JL, Byers PH, Andelfinger G, Siu V, Lauzon J, Fernandez BA, McMillin M, Scott RH, Racher H, Majewski J, Nickerson DA, Shendure J, Bamshad MJ, Parboosingh JS FORGE Canada Consortium. Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am J Hum Genet. 2012;90:925–933. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
