

16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice
- PMID: 26120470
- PMCID: PMC4482049
- DOI: 10.1186/s40168-015-0087-4
16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice
Abstract
Background: Characterisation of the bacterial composition of the gut microbiota is increasingly carried out with a view to establish the role of different bacterial species in causation or prevention of disease. It is thus essential that the methods used to determine the microbial composition are robust. Here, several widely used molecular techniques were compared to establish the optimal methods to assess the bacterial composition in faecal samples from babies, before weaning.
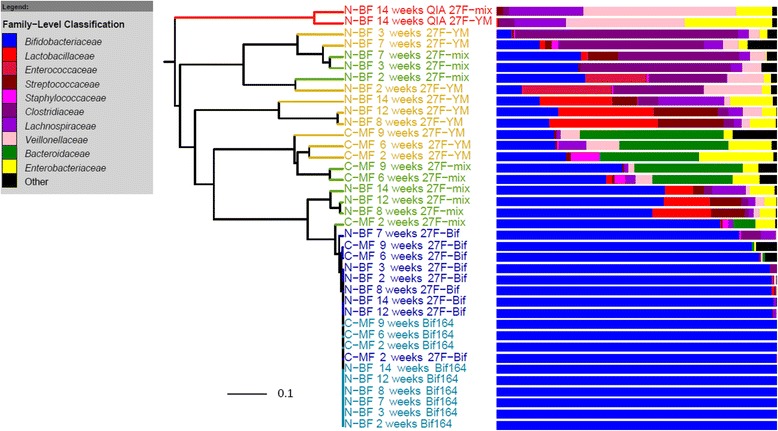
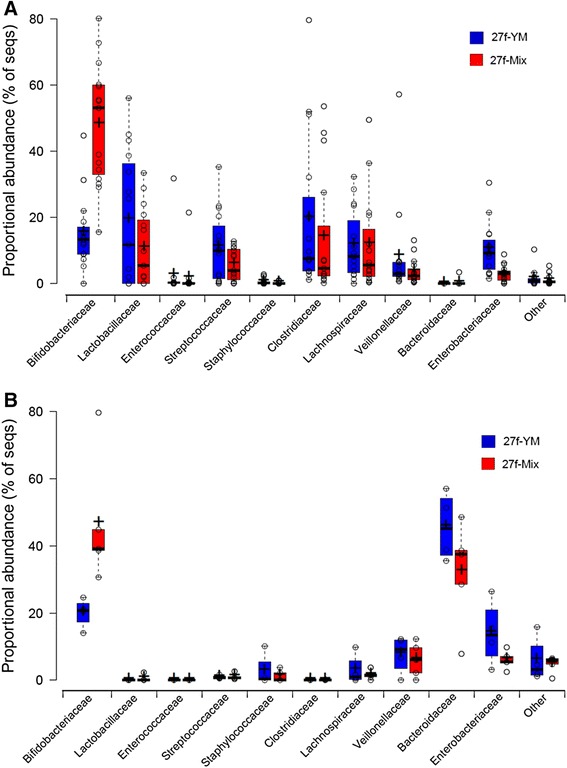
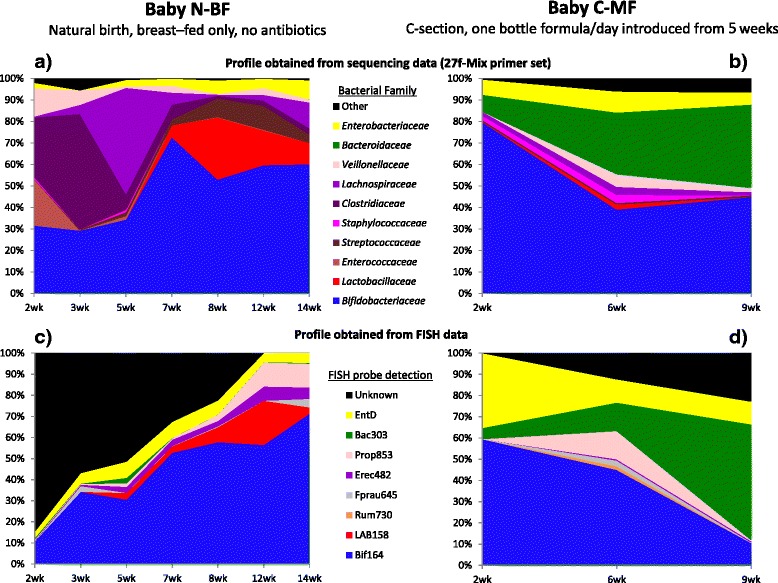
Results: The bacterial community profile detected in the faeces of infants is highly dependent on the methodology used. Bifidobacteria were the most abundant bacteria detected at 6 weeks in faeces from two initially breast-fed babies using fluorescent in situ hybridisation (FISH), in agreement with data from previous culture-based studies. Using the 16S rRNA gene sequencing approach, however, we found that the detection of bifidobacteria in particular crucially depended on the optimisation of the DNA extraction method, and the choice of primers used to amplify the V1-V3 regions of 16S rRNA genes prior to subsequent sequence analysis. Bifidobacteria were only well represented among amplified 16S rRNA gene sequences when mechanical disruption (bead-beating) procedures for DNA extraction were employed together with optimised "universal" PCR primers. These primers incorporate degenerate bases at positions where mismatches to bifidobacteria and other bacterial taxa occur. The use of a DNA extraction kit with no bead-beating step resulted in a complete absence of bifidobacteria in the sequence data, even when using the optimised primers.
Conclusions: This work emphasises the importance of sample processing methodology to downstream sequencing results and illustrates the value of employing multiple approaches for determining microbiota composition.
Keywords: 16S rRNA gene sequencing; Bifidobacteria; FISH; Infant; Intestinal microbiota.
Figures
References
-
- Mulder IE, Schmidt B, Lewis M, Delday M, Stokes CR, Bailey M, Aminov RI, Gill BP, Pluske JR, Mayer C-D, Kelly D. Restricting microbial exposure in early life negates the immune benefits associated with gut colonization in environments of high microbial diversity. PLoS ONE 2011, 6(12); doi:10.1371/journal.pone.0028279. - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
