

Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease
- PMID: 26122708
- PMCID: PMC4659505
- DOI: 10.1016/j.freeradbiomed.2015.06.021
Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease
Abstract
Nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) regulates the basal and stress-inducible expression of a battery of genes encoding key components of the glutathione-based and thioredoxin-based antioxidant systems, as well as aldo-keto reductase, glutathione S-transferase, and
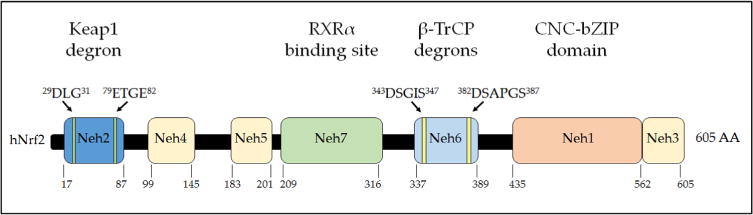
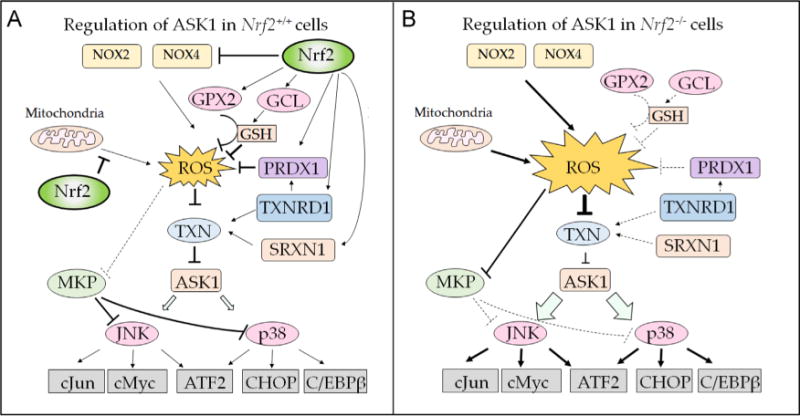
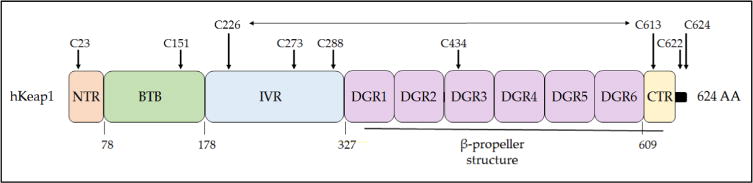
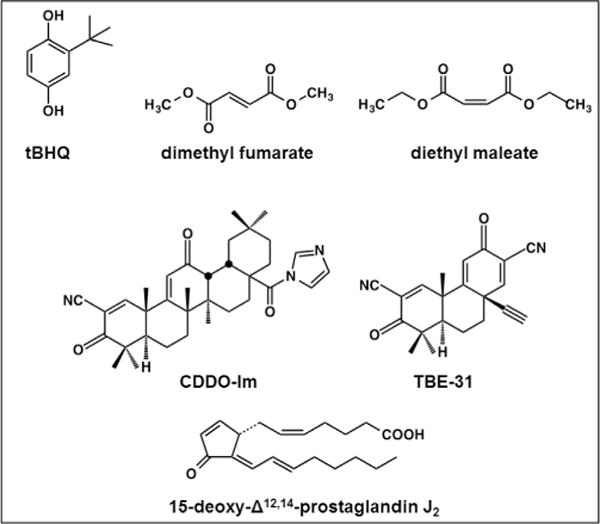
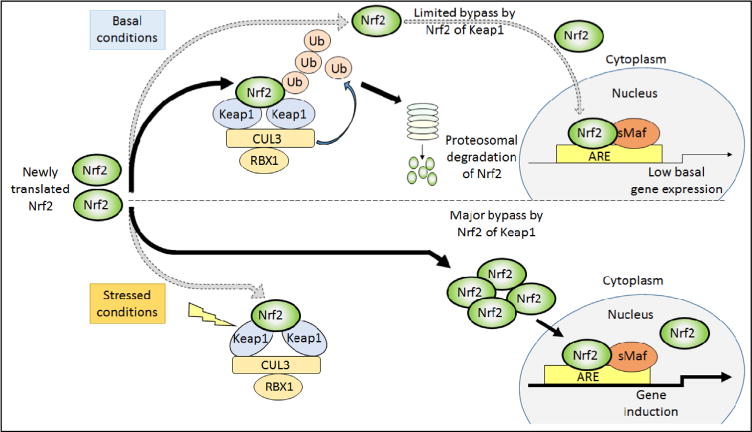
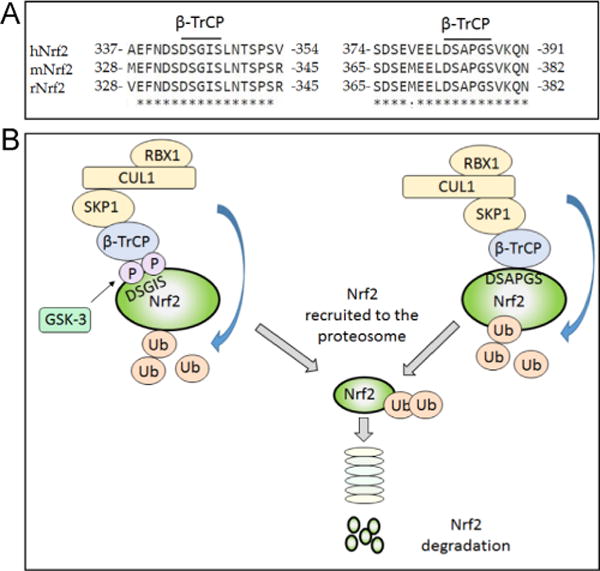
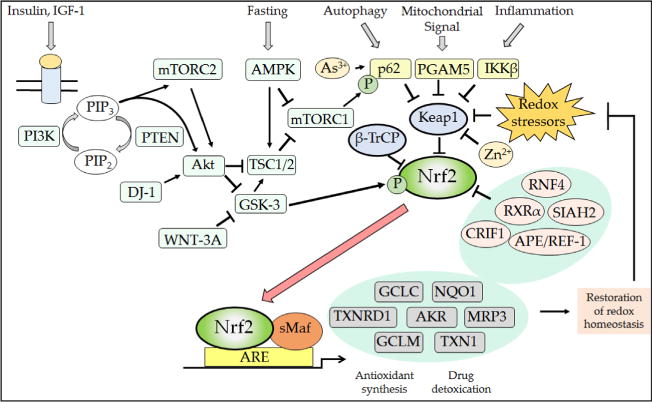
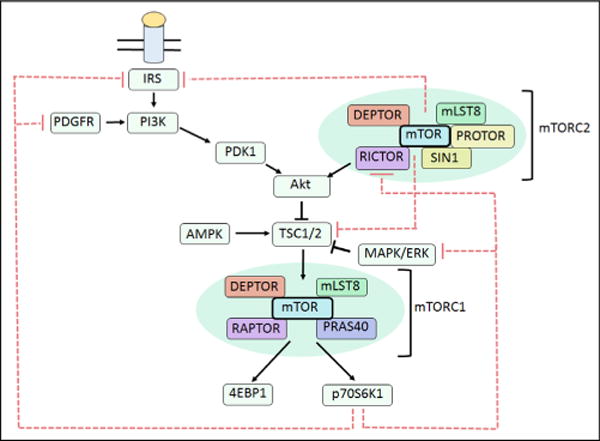
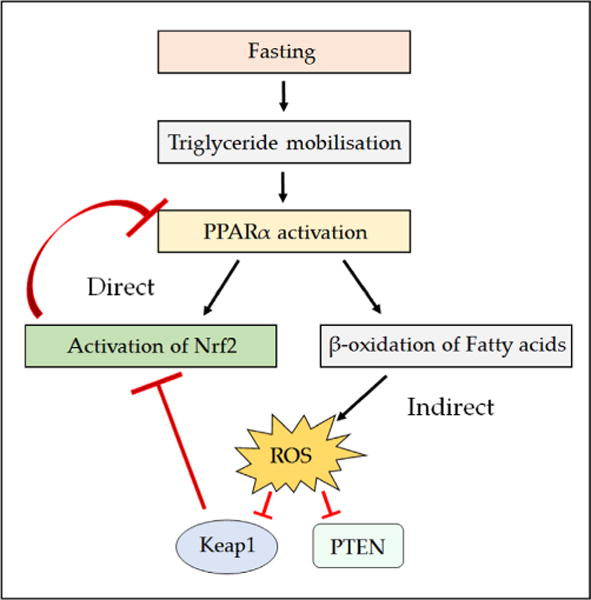
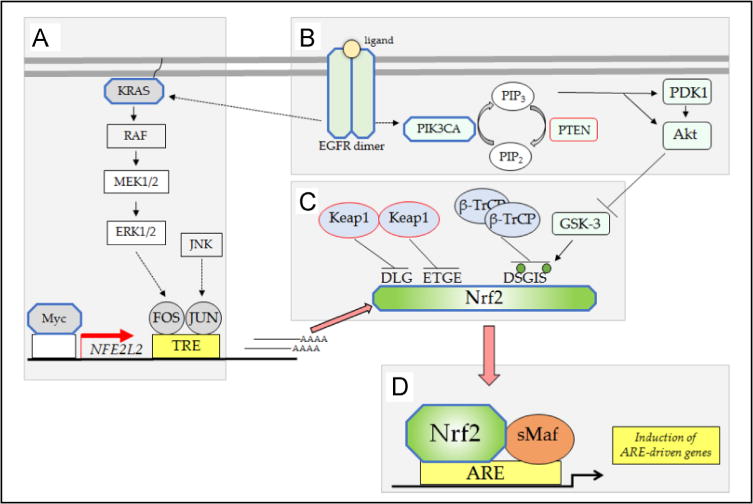
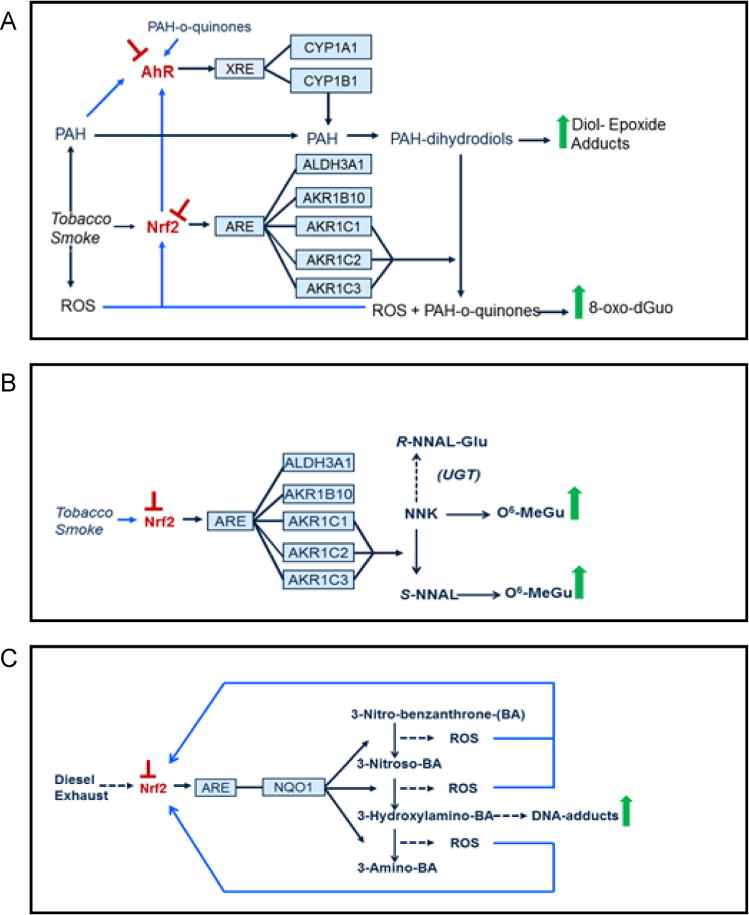
Nad(p)h: quinone oxidoreductase-1 drug-metabolizing isoenzymes along with multidrug-resistance-associated efflux pumps. It therefore plays a pivotal role in both intrinsic resistance and cellular adaptation to reactive oxygen species (ROS) and xenobiotics. Activation of Nrf2 can, however, serve as a double-edged sword because some of the genes it induces may contribute to chemical carcinogenesis by promoting futile redox cycling of polycyclic aromatic hydrocarbon metabolites or confer resistance to chemotherapeutic drugs by increasing the expression of efflux pumps, suggesting its cytoprotective effects will vary in a context-specific fashion. In addition to cytoprotection, Nrf2 also controls genes involved in intermediary metabolism, positively regulating those involved in NADPH generation, purine biosynthesis, and the β-oxidation of fatty acids, while suppressing those involved in lipogenesis and gluconeogenesis. Nrf2 is subject to regulation at multiple levels. Its ability to orchestrate adaptation to oxidants and electrophiles is due principally to stress-stimulated modification of thiols within one of its repressors, the Kelch-like ECH-associated protein 1 (Keap1), which is present in the cullin-3 RING ubiquitin ligase (CRL) complex CRLKeap1. Thus modification of Cys residues in Keap1 blocks CRLKeap1 activity, allowing newly translated Nrf2 to accumulate rapidly and induce its target genes. The ability of Keap1 to repress Nrf2 can be attenuated by p62/sequestosome-1 in a mechanistic target of rapamycin complex 1 (mTORC1)-dependent manner, thereby allowing refeeding after fasting to increase Nrf2-target gene expression. In parallel with repression by Keap1, Nrf2 is also repressed by β-transducin repeat-containing protein (β-TrCP), present in the Skp1-cullin-1-F-box protein (SCF) ubiquitin ligase complex SCFβ-TrCP. The ability of SCFβ-TrCP to suppress Nrf2 activity is itself enhanced by prior phosphorylation of the transcription factor by glycogen synthase kinase-3 (GSK-3) through formation of a DSGIS-containing phosphodegron. However, formation of the phosphodegron in Nrf2 by GSK-3 is inhibited by stimuli that activate protein kinase B (PKB)/Akt. In particular, PKB/Akt activity can be increased by phosphoinositide 3-kinase and mTORC2, thereby providing an explanation of why antioxidant-responsive element-driven genes are induced by growth factors and nutrients. Thus Nrf2 activity is tightly controlled via CRLKeap1 and SCFβ-TrCP by oxidative stress and energy-based signals, allowing it to mediate adaptive responses that restore redox homeostasis and modulate intermediary metabolism. Based on the fact that Nrf2 influences multiple biochemical pathways in both positive and negative ways, it is likely its dose-response curve, in terms of susceptibility to certain degenerative disease, is U-shaped. Specifically, too little Nrf2 activity will lead to loss of cytoprotection, diminished antioxidant capacity, and lowered β-oxidation of fatty acids, while conversely also exhibiting heightened sensitivity to ROS-based signaling that involves receptor tyrosine kinases and apoptosis signal-regulating kinase-1. By contrast, too much Nrf2 activity disturbs the homeostatic balance in favor of reduction, and so may have deleterious consequences including overproduction of reduced glutathione and NADPH, the blunting of ROS-based signal transduction, epithelial cell hyperplasia, and failure of certain cell types to differentiate correctly. We discuss the basis of a putative U-shaped Nrf2 dose-response curve in terms of potentially competing processes relevant to different stages of tumorigenesis.
Keywords: Free radicals; GSK-3; Glutathione; Inflammation; Keap1; Lipid metabolism; Nrf2; Nutrient supply; Reactive oxygen species; Thioredoxin; aldo-keto reductase (AKR); β-TrCP.
Copyright © 2015. Published by Elsevier Inc.
Figures
References
-
- Halliwell B. Biochemistry of oxidative stress. Biochem Soc Trans. 2007;35:1147–1150. - PubMed
-
- Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. - PubMed
-
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- C4909/A13786/CRUK_/Cancer Research UK/United Kingdom
- P30 CA006973/CA/NCI NIH HHS/United States
- BB/L01923X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- P30 ES013508/ES/NIEHS NIH HHS/United States
- BB/L01405X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
