

Review
doi: 10.1038/ncb3184.
Proteostasis control by the unfolded protein response
Affiliations
- PMID: 26123108
- PMCID: PMC5546321
- DOI: 10.1038/ncb3184
Item in Clipboard
Review
Proteostasis control by the unfolded protein response
Nat Cell Biol.
2015 Jul.
Erratum in
-
Erratum: Proteostasis control by the unfolded protein response.Nat Cell Biol. 2015 Aug;17(8):1088. doi: 10.1038/ncb3221. Nat Cell Biol. 2015. PMID: 26239530 Free PMC article.
Abstract
Stress induced by accumulation of misfolded proteins in the endoplasmic reticulum is observed in many physiological and pathological conditions. To cope with endoplasmic reticulum stress, cells activate the unfolded protein response, a dynamic signalling network that orchestrates the recovery of homeostasis or triggers apoptosis, depending on the level of damage. Here we provide an overview of recent insights into the mechanisms that cells employ to maintain proteostasis and how the unfolded protein response determines cell fate under endoplasmic reticulum stress.
Figures
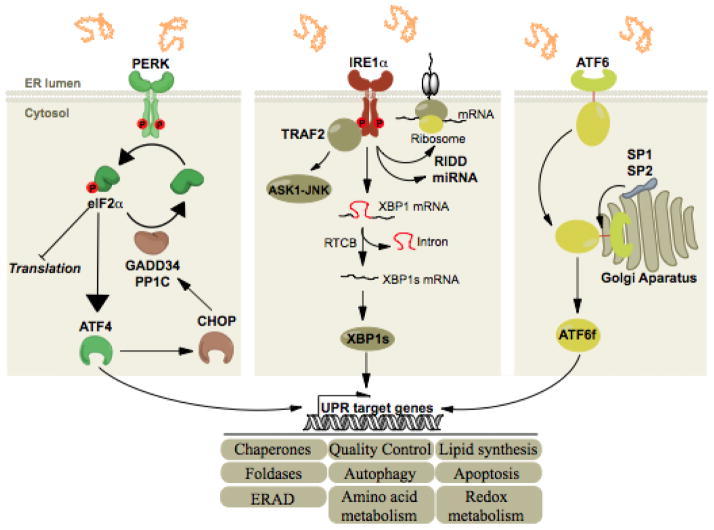
All three ER stress sensors (PERK, IRE1α, ATF6) initially activate signaling events that increase protein-folding capacity and reduce protein load on the ER. These transcriptional and translational outputs tend to re-establish protein-folding homeostasis in the ER and promote cell survival. PERK phosphorylates eIF2alpha, which in turn shut down global translation and in the mean time increases the expression of the transcription factor ATF4. The latter induces the transcription of select genes whose functions are to restore proteostasis (grey boxes) and of CHOP, itself inducing the transcription of GADD34, a regulatory subunit of PP1C. This creates a feedback mechanism leading to the dephosphorylation of eIF2alpha and translation is reinitiated. IRE1α signals through i) the recruitment of TRAF2 leading the activation of the ASK1-JNK cascade and ii) through its RNase via the splicing of XBP1 mRNA or the degradation of RNAs, thereby regulating gene expression at transcriptional and post-transcriptional levels. Finally, upon ER stress, ATF6 is exported from the ER to the Golgi complex where it is cleaved by the proteases S1P and S2P, releasing its cytosolic domain which is a potent transcription factor.
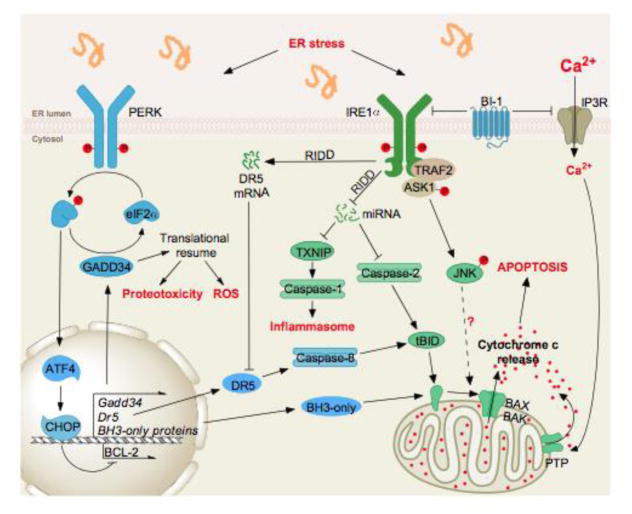
When exposed to chronically high levels of ER stress, PERK and IRE1α both have multiple signaling outputs that lead to cell dysfunction, activation of the inflammasome, and apoptosis. Among the ATF4 targets downstream of PERK (see Figure 1) are also found genes whose products are involved in the control of cell death (DR5 or BH3-only) through the activation of signaling pathways from the plasma membrane or the mitochondria. Similarly, IRE1α signals through the JNK and mRNA/miRNA degradation pathways to control cell survival outputs. Interestingly, specific cross-talks between IRE1α and PERK signaling, notably at the level of DR5, that tightly control cell fate.
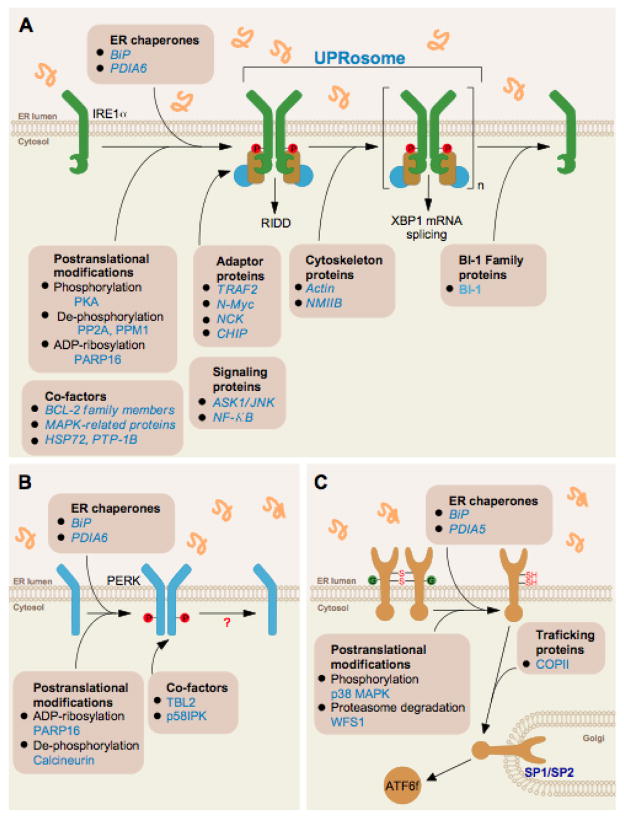
(A) Fine-tuning IRE1 signaling through the dissociation of ER chaperones in the activation phase, recruitment of the UPRosome that controls signaling outputs and inactivation phase. (B) Fine-tuning PERK activation through the dissociation of BiP in the activation phase and the control of phosphorylation and ADP-ribosylation. (C) ATF6α activation depends on its N-glycosylation and redox status in addition to the dissociation from BiP to allow egress from the ER and proteolytic cleavage in the Golgi apparatus by the S1P/S2P proteases. In addition, ATF6α protein stability plays a key role in its signaling outputs.
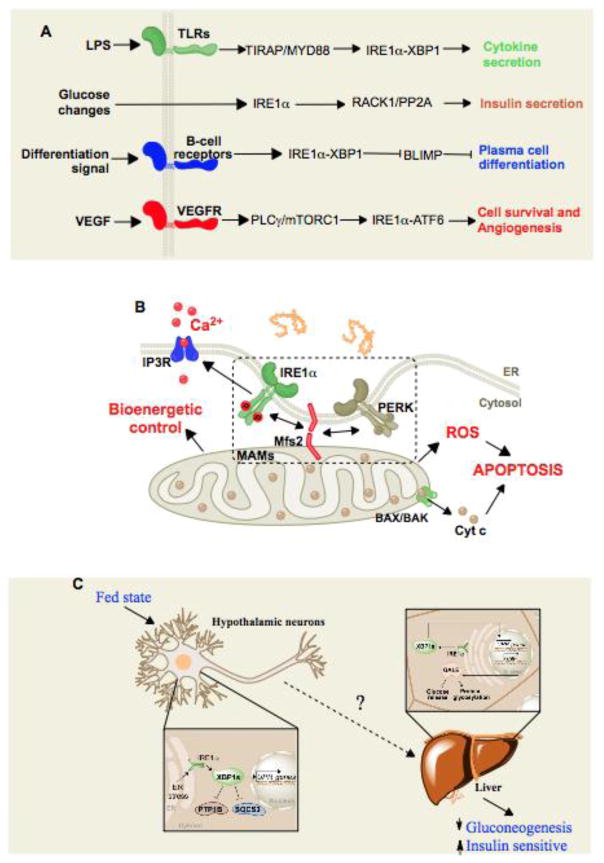
(A) Signaling (LPS, glucose, differentiation, VEGF)-dependent and protein misfolding-independent activation of the canonical UPR sensors. (B) Control of mitochondrial function by UPR signaling at the ER-mitochondrion interface. (C) Cell-nonautonomous UPR activation. ER stress is triggered in neurons that subsequently prompt the activation of select UPR signals in the non-stressed liver through yet uncharacterized mediators.
References
-
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. - PubMed
-
- Schubert U, et al. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. - PubMed
-
- Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
