

De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy
- PMID: 26125038
- PMCID: PMC4479523
- DOI: 10.1002/acn3.198
De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy
Abstract
Objective: To determine the cause and course of a novel syndrome with progressive encephalopathy and brain atrophy in children.
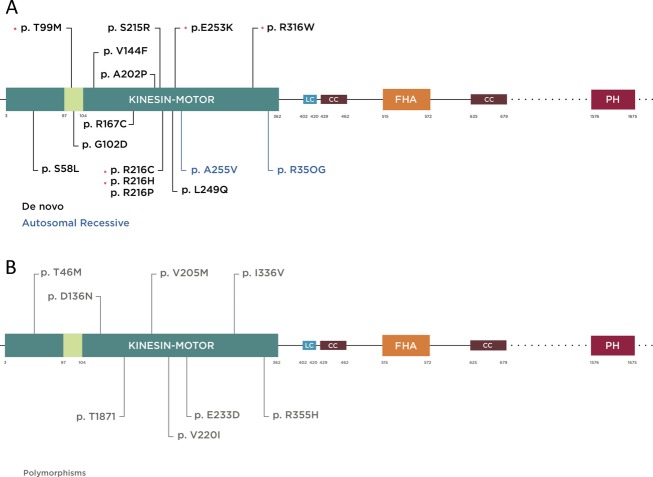
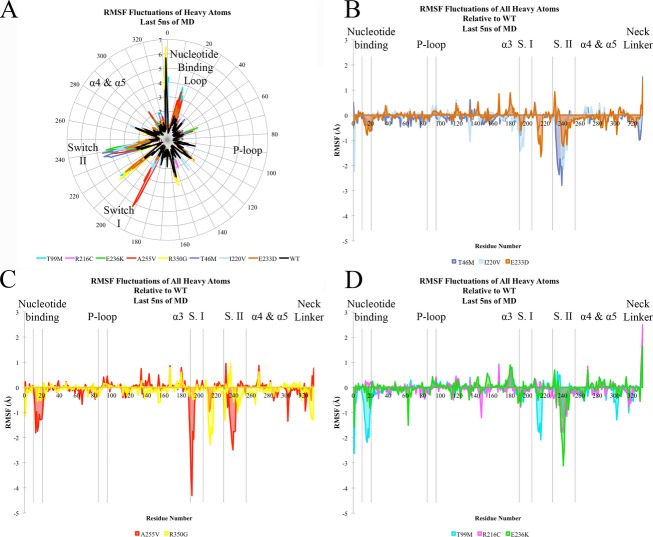
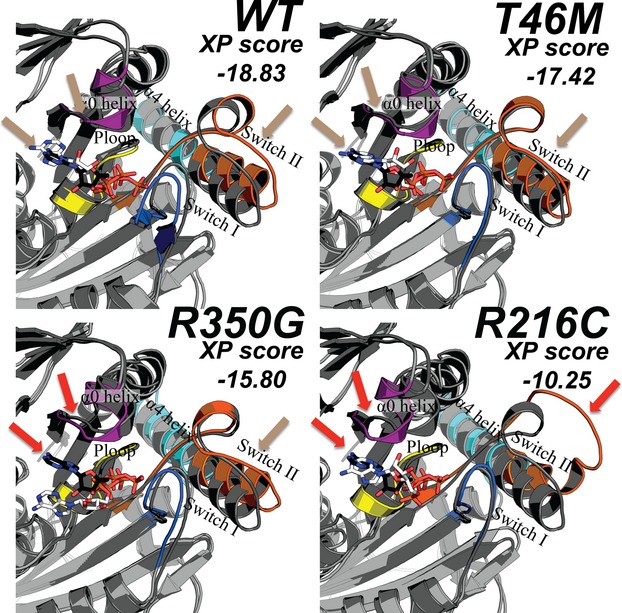
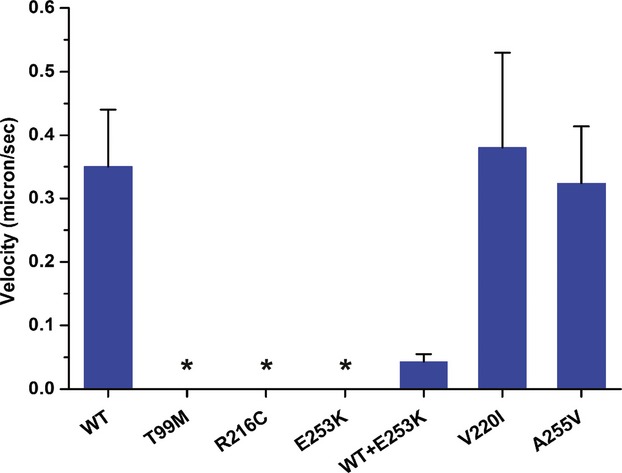
Methods: Clinical whole-exome sequencing was performed for global developmental delay and intellectual disability; some patients also had spastic paraparesis and evidence of clinical regression. Six patients were identified with de novo missense mutations in the kinesin gene KIF1A. The predicted functional disruption of these mutations was assessed in silico to compare the calculated conformational flexibility and estimated efficiency of ATP binding to kinesin motor domains of wild-type (WT) versus mutant alleles. Additionally, an in vitro microtubule gliding assay was performed to assess the effects of de novo dominant, inherited recessive, and polymorphic variants on KIF1A motor function.
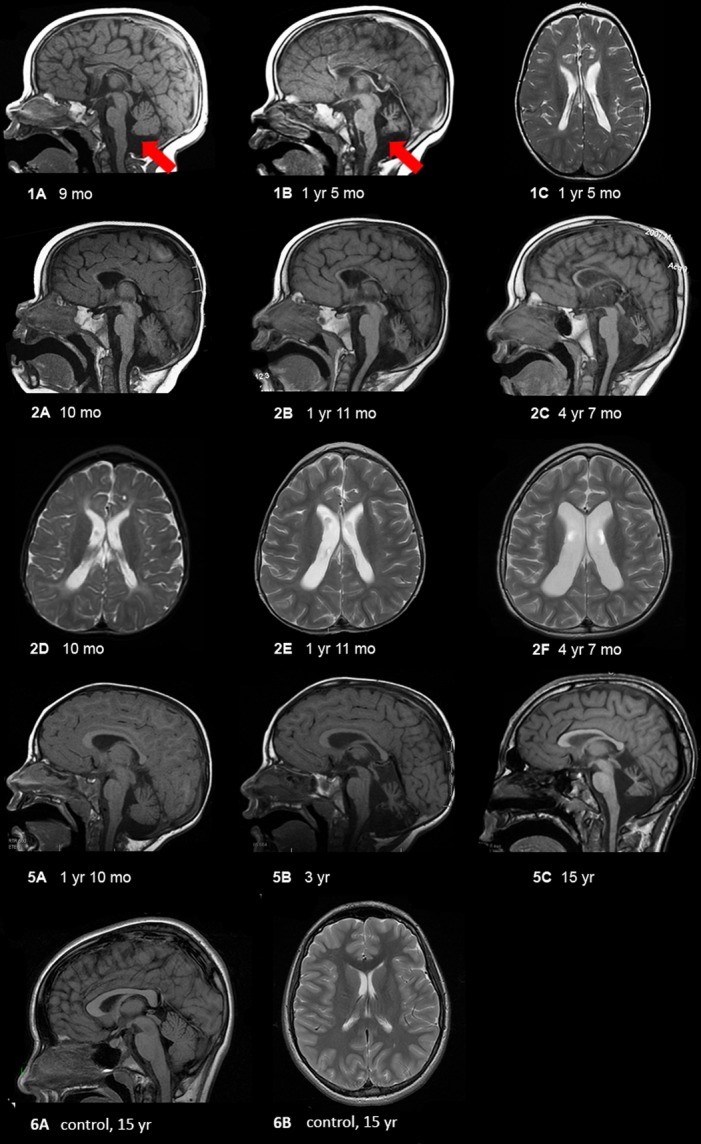
Results: All six subjects had severe developmental delay, hypotonia, and varying degrees of hyperreflexia and spastic paraparesis. Microcephaly, cortical visual impairment, optic neuropathy, peripheral neuropathy, ataxia, epilepsy, and movement disorders were also observed. All six patients had a degenerative neurologic course with progressive cerebral and cerebellar atrophy seen on sequential magnetic resonance imaging scans. Computational modeling of mutant protein structures when compared to WT kinesin showed substantial differences in conformational flexibility and ATP-binding efficiency. The de novo KIF1A mutants were nonmotile in the microtubule gliding assay.
Interpretation: De novo mutations in KIF1A cause a degenerative neurologic syndrome with brain atrophy. Computational and in vitro assays differentiate the severity of dominant de novo heterozygous versus inherited recessive KIF1A mutations. The profound effect de novo mutations have on axonal transport is likely related to the cause of progressive neurologic impairment in these patients.
Figures
References
-
- Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
