

Porphyria Diagnostics-Part 1: A Brief Overview of the Porphyrias
- PMID: 26132003
- PMCID: PMC4640448
- DOI: 10.1002/0471142905.hg1720s86
Porphyria Diagnostics-Part 1: A Brief Overview of the Porphyrias
Abstract
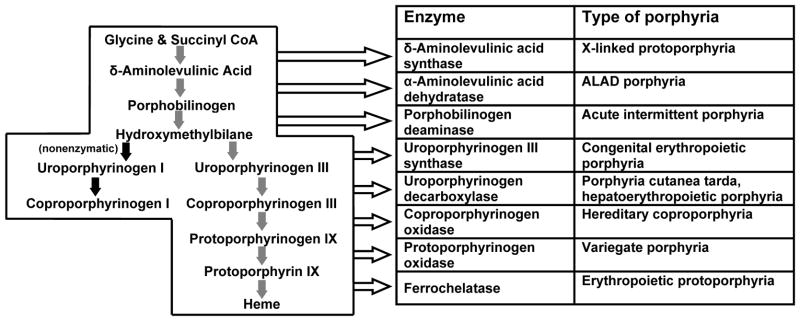
Porphyria diseases are a group of metabolic disorders caused by abnormal functioning of heme biosynthesis enzymes and characterized by excessive accumulation and excretion of porphyrins and their precursors. Precisely which of these chemicals builds up depends on the type of porphyria. Porphyria is not a single disease but a group of nine disorders: acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), δ-aminolevulinic acid dehydratase deficiency porphyria (ADP), porphyria cutanea tarda (PCT), hepatoerythropoietic porphyria (HEP), congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLP). Each porphyria results from overproduction of heme precursors secondary to partial deficiency or, in XLP, increased activity of one of the enzymes of heme biosynthesis. Taken together, all forms of porphyria afflict fewer than 200,000 people in the United States. Based on European studies, the most common porphyria, PCT, has a prevalence of 1 in 10,000, the most common acute porphyria, AlP, has a prevalence of ∼1 in 20,000, and the most common erythropoietic porphyria, EPP, is estimated at 1 in 50,000 to 75,000. CEP is extremely rare, with prevalence estimates of 1 in 1,000,000 or less. Only six cases of ADP are documented. The current porphyria literature is very exhaustive and a brief overview of porphyria diseases is essential in order for the reader to better appreciate the relevance of this area of research prior to undertaking biochemical diagnostics procedures. This unit summarizes the current knowledge on the classification, clinical features, etiology, pathogenesis, and genetics of porphyria diseases.
Keywords: Porphyria; clinical features; etiology; overview; pathogenesis.
Copyright © 2015 John Wiley & Sons, Inc.
Figures
References
-
- Aarsand AK, Boman H, Sandberg S. Familial and sporadic porphyria cutanea tarda: characterization and diagnostic strategies. Clin Chem. 2009;55:795–803. - PubMed
-
- Akagi R, Kato N, Inoue R, Anderson KE, Jaffe EK, Sassa S. Delta-Aminolevulinate dehydratase (ALAD) porphyria: the first case in North America with two novel ALAD mutations. Mol Genet Metab. 2006;87:329–36. - PubMed
-
- Albers JW, Robertson WC, Jr, Daube JR. Electrodiagnostic findings in acute porphyric neuropathy. Muscle Nerve. 1978;1(4):292–6. - PubMed
-
- Anderson KE. Approaches to treatment and prevention of human porphyrias. In: Kadish KK, Smith KM, Guilard R, editors. The Porphyrin Handbook, Vol. 14: Medical Aspects of Porphyrins. Academic Press; San Diego, CA: 2003. pp. 247–283.
-
- Anderson KE. Chapter 152. Clinical and laboratory diagnosis of the porphyrias. In: Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. Handbook of porphyrin science with applications in chemistry, physics, materials science, engineering, biology, and medicine, Vol. 29: Porphyrias and sideroblastic anemias. World Scientific Publishing Co., Private Limited; Hackensack, NJ 07601, USA: 2014. pp. 370–406.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
