

The Sick and the Weak: Neuropathies/Myopathies in the Critically Ill
- PMID: 26133937
- PMCID: PMC4491544
- DOI: 10.1152/physrev.00028.2014
The Sick and the Weak: Neuropathies/Myopathies in the Critically Ill
Abstract
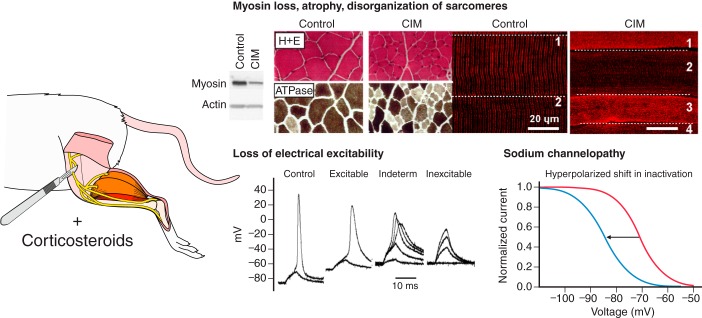
Critical illness polyneuropathies (CIP) and myopathies (CIM) are common complications of critical illness. Several weakness syndromes are summarized under the term intensive care unit-acquired weakness (ICUAW). We propose a classification of different ICUAW forms (CIM, CIP, sepsis-induced, steroid-denervation myopathy) and pathophysiological mechanisms from clinical and animal model data. Triggers include sepsis, mechanical ventilation, muscle unloading, steroid treatment, or denervation. Some ICUAW forms require stringent diagnostic features; CIM is marked by membrane hypoexcitability, severe atrophy, preferential myosin loss, ultrastructural alterations, and inadequate autophagy activation while myopathies in pure sepsis do not reproduce marked myosin loss. Reduced membrane excitability results from depolarization and ion channel dysfunction. Mitochondrial dysfunction contributes to energy-dependent processes. Ubiquitin proteasome and calpain activation trigger muscle proteolysis and atrophy while protein synthesis is impaired. Myosin loss is more pronounced than actin loss in CIM. Protein quality control is altered by inadequate autophagy. Ca(2+) dysregulation is present through altered Ca(2+) homeostasis. We highlight clinical hallmarks, trigger factors, and potential mechanisms from human studies and animal models that allow separation of risk factors that may trigger distinct mechanisms contributing to weakness. During critical illness, altered inflammatory (cytokines) and metabolic pathways deteriorate muscle function. ICUAW prevention/treatment is limited, e.g., tight glycemic control, delaying nutrition, and early mobilization. Future challenges include identification of primary/secondary events during the time course of critical illness, the interplay between membrane excitability, bioenergetic failure and differential proteolysis, and finding new therapeutic targets by help of tailored animal models.
Copyright © 2015 the American Physiological Society.
Figures















References
-
- Aare S, Ochala J, Norman HS, Radell P, Eriksson LI, Goransson H, Chen YW, Hoffman EP, Larsson L. Mechanisms underlying the sparing of masticatory versus limb muscle function in an experimental critical illness model. Physiol Genomics 43: 1334–1350, 2011. - PubMed
-
- Aare S, Radell P, Eriksson LI, Chen YW, Hoffman EP, Larsson L. The role of sepsis in the development of limb muscle weakness in a porcine intensive care unit model. Physiol Genomics 44: 865–877, 2012. - PubMed
-
- Abdellaoui A, Prefaut C, Gouzi F, Couillard A, Coisy-Quivy M, Hugon G, Molinari N, Lafontaine T, Jonquet O, Laoudj-Chenivesse D, Hayot M. Skeletal muscle effects of electrostimulation after COPD exacerbation: a pilot study. Eur Respir J 38: 781–788, 2011. - PubMed
-
- Adams V, Nehrhoff B, Späte U, Linke A, Schulze PC, Baur A, Gielen S, Hambrecht R, Chuler G. Induction of iNOS expression in skeletal muscle by IL-1beta and NFkappaB activation. An in vitro and in vivo study. Cardiovasc Res 54: 95–104, 2002. - PubMed
-
- Adhihetty PJ, O'Leary MFN, Chabi B, Wicks KL, Hood DA. Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J Appl Physiol 102: 1143–1151, 2007. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous