

Host Cell Plasma Membrane Phosphatidylserine Regulates the Assembly and Budding of Ebola Virus
- PMID: 26136573
- PMCID: PMC4542376
- DOI: 10.1128/JVI.01087-15
Host Cell Plasma Membrane Phosphatidylserine Regulates the Assembly and Budding of Ebola Virus
Abstract
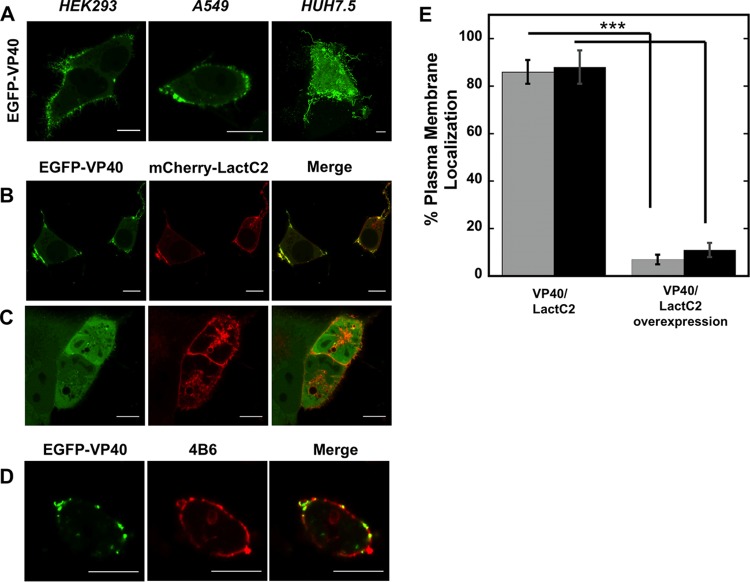
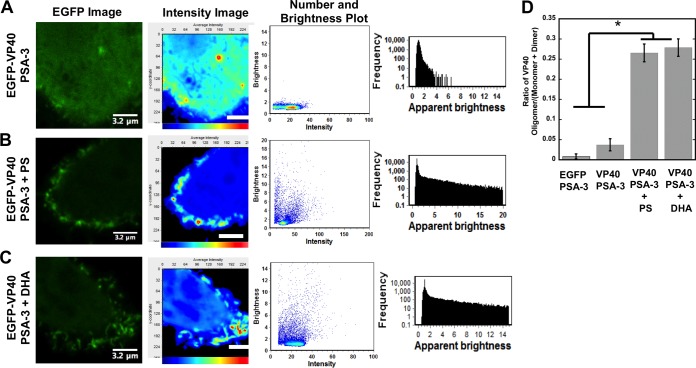
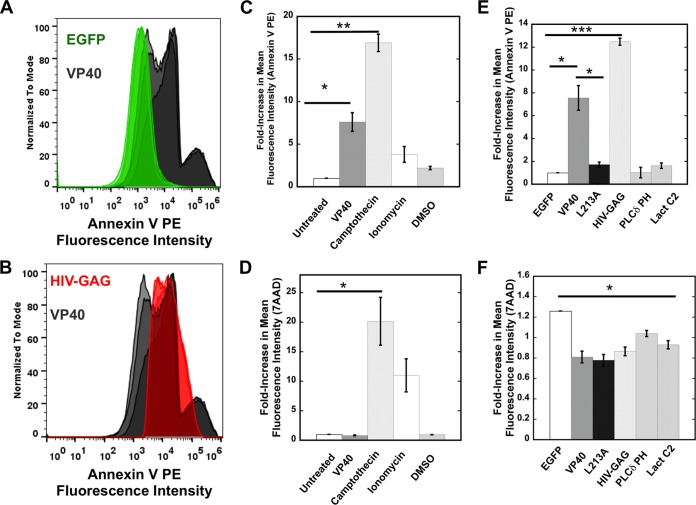
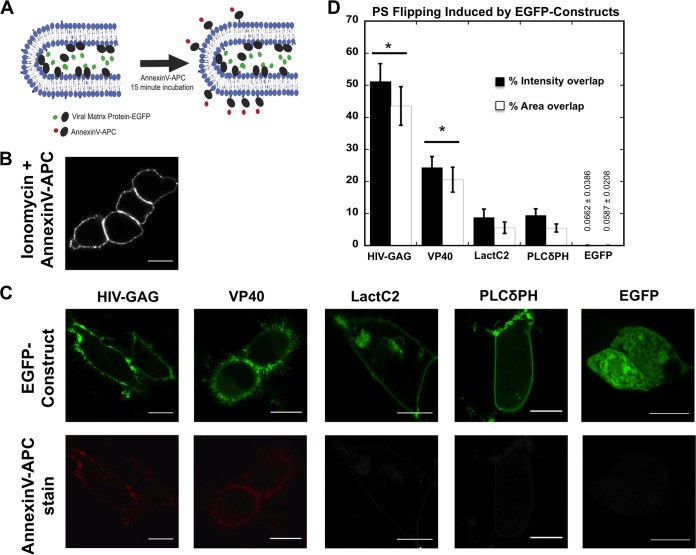
Lipid-enveloped viruses replicate and bud from the host cell where they acquire their lipid coat. Ebola virus, which buds from the plasma membrane of the host cell, causes viral hemorrhagic fever and has a high fatality rate. To date, little has been known about how budding and egress of Ebola virus are mediated at the plasma membrane. We have found that the lipid phosphatidylserine (PS) regulates the assembly of Ebola virus matrix protein VP40. VP40 binds PS-containing membranes with nanomolar affinity, and binding of PS regulates VP40 localization and oligomerization on the plasma membrane inner leaflet. Further, alteration of PS levels in mammalian cells inhibits assembly and egress of VP40. Notably, interactions of VP40 with the plasma membrane induced exposure of PS on the outer leaflet of the plasma membrane at sites of egress, whereas PS is typically found only on the inner leaflet. Taking the data together, we present a model accounting for the role of plasma membrane PS in assembly of Ebola virus-like particles.
Importance: The lipid-enveloped Ebola virus causes severe infection with a high mortality rate and currently lacks FDA-approved therapeutics or vaccines. Ebola virus harbors just seven genes in its genome, and there is a critical requirement for acquisition of its lipid envelope from the plasma membrane of the human cell that it infects during the replication process. There is, however, a dearth of information available on the required contents of this envelope for egress and subsequent attachment and entry. Here we demonstrate that plasma membrane phosphatidylserine is critical for Ebola virus budding from the host cell plasma membrane. This report, to our knowledge, is the first to highlight the role of lipids in human cell membranes in the Ebola virus replication cycle and draws a clear link between selective binding and transport of a lipid across the membrane of the human cell and use of that lipid for subsequent viral entry.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
