

Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome
- PMID: 26137199
- PMCID: PMC4484546
- DOI: 10.1016/j.csbj.2015.06.001
Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome
Abstract
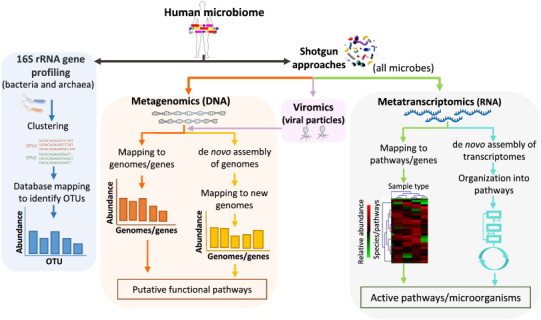
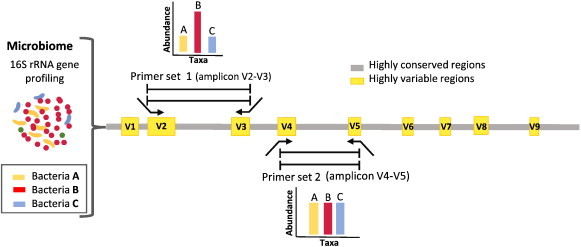
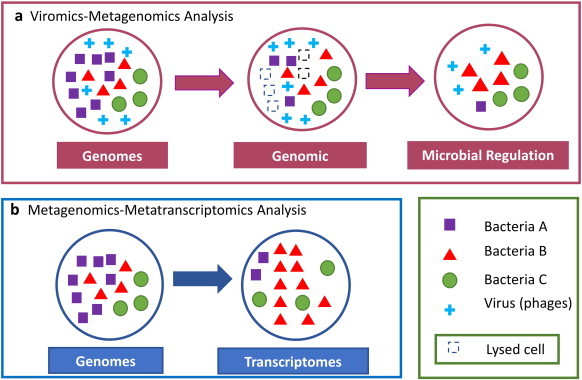
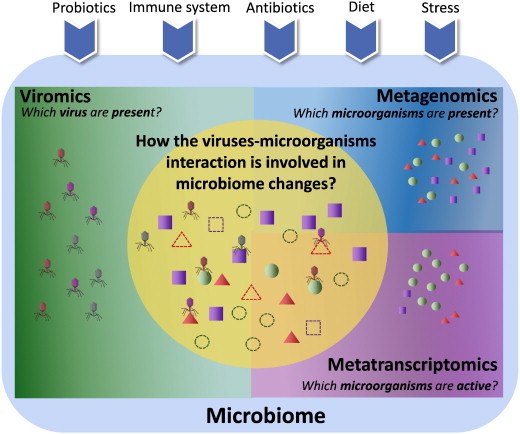
The advances in experimental methods and the development of high performance bioinformatic tools have substantially improved our understanding of microbial communities associated with human niches. Many studies have documented that changes in microbial abundance and composition of the human microbiome is associated with human health and diseased state. The majority of research on human microbiome is typically focused in the analysis of one level of biological information, i.e., metagenomics or metatranscriptomics. In this review, we describe some of the different experimental and bioinformatic strategies applied to analyze the 16S rRNA gene profiling and shotgun sequencing data of the human microbiome. We also discuss how some of the recent insights in the combination of metagenomics, metatranscriptomics and viromics can provide more detailed description on the interactions between microorganisms and viruses in oral and gut microbiomes. Recent studies on viromics have begun to gain importance due to the potential involvement of viruses in microbial dysbiosis. In addition, metatranscriptomic combined with metagenomic analysis have shown that a substantial fraction of microbial transcripts can be differentially regulated relative to their microbial genomic abundances. Thus, understanding the molecular interactions in the microbiome using the combination of metagenomics, metatranscriptomics and viromics is one of the main challenges towards a system level understanding of human microbiome.
Keywords: Bioinformatics; Human microbiome; Metagenomics; Metatranscriptomics; Systems-level; Viromics.
Figures
References
-
- Hooper L.V., Gordon J.I. Commensal host–bacterial relationships in the gut. Science. 2001;292(5519):1115–1118. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources