

Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis
- PMID: 26137539
- PMCID: PMC4484512
- DOI: 10.1016/j.ebiom.2014.12.005
Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis
Abstract
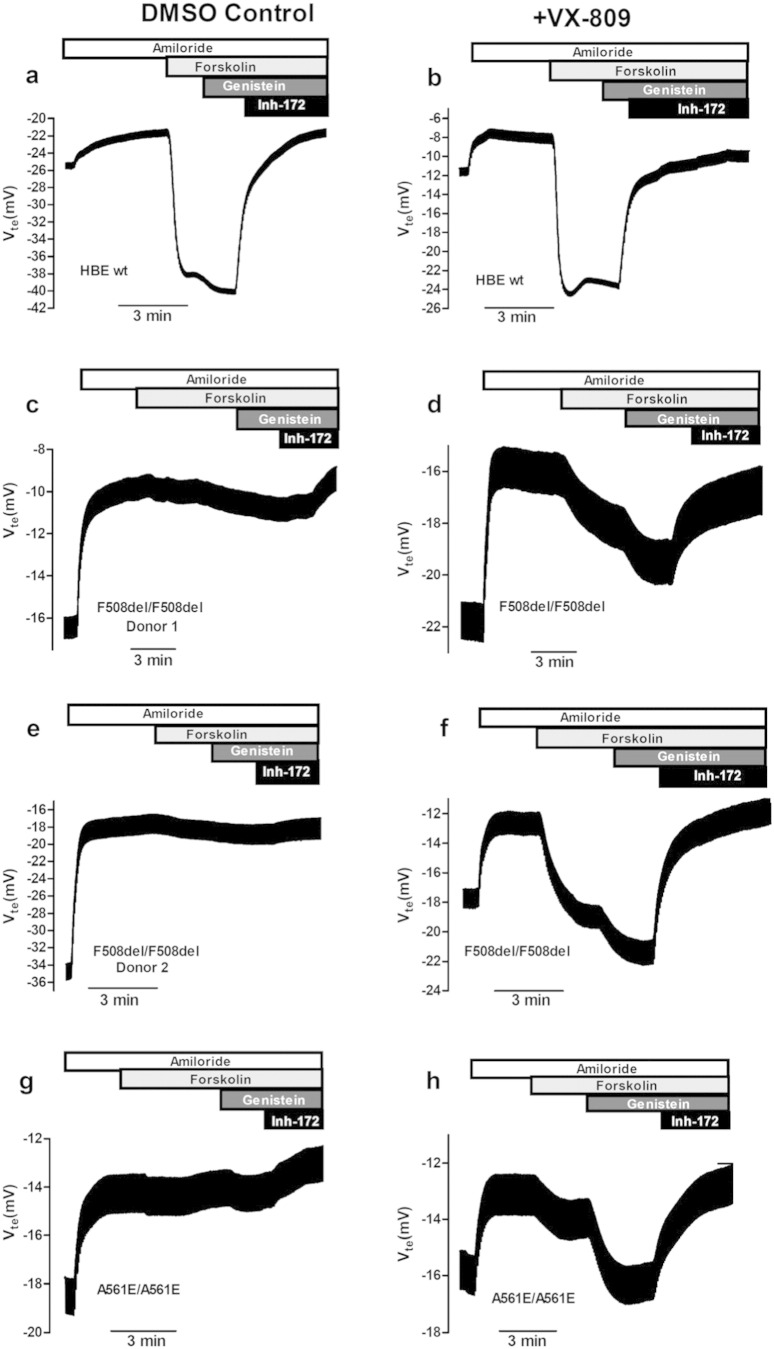
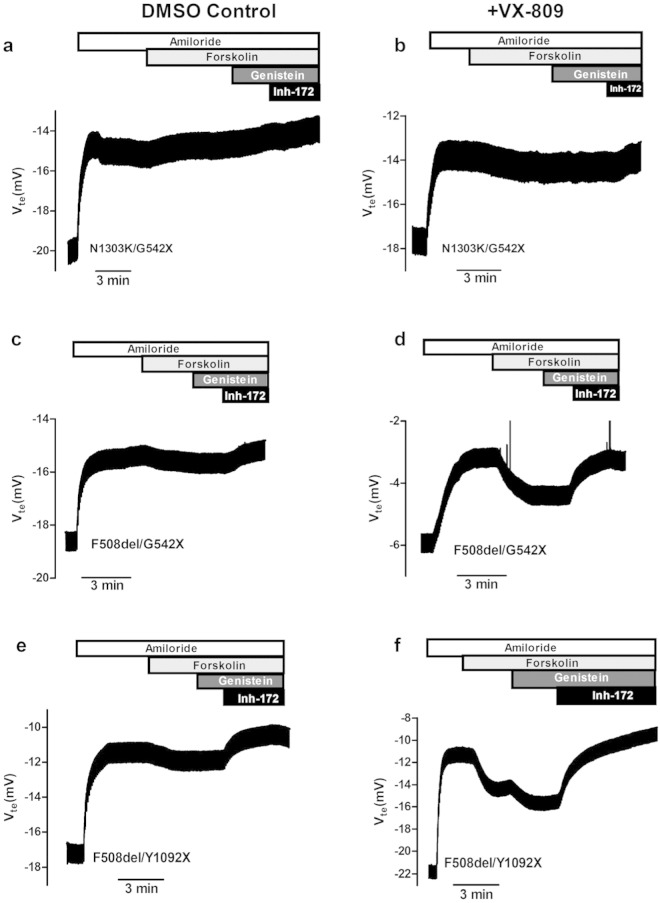
Background: The best investigational drug to treat cystic fibrosis (CF) patients with the most common CF-causing mutation (F508del) is VX-809 (lumacaftor) which recently succeeded in Phase III clinical trial in combination with ivacaftor. This corrector rescues F508del-CFTR from its abnormal intracellular localization to the cell surface, a traffic defect shared by all Class II CFTR mutants. Our goal here is to test the efficacy of lumacaftor in other Class II mutants in primary human bronchial epithelial (HBE) cells derived from CF patients.
Methods: The effect of lumacaftor was investigated in primary HBE cells from non-CF and CF patients with F508del/F508del, A561E/A561E, N1303K/G542X, F508del/G542X and F508del/Y1092X genotypes by measurements of Forskolin plus Genistein-inducible equivalent short-circuit current (Ieq-SC-Fsk + Gen) in perfused open-circuit Ussing chambers. Efficacy of corrector C18 was also assessed on A561E/A561E and F508del/F508del cells.
Results: Our data indicate that A561E (when present in both alleles) responds positively to lumacaftor treatment at equivalent efficacy of F508del in primary HBE cells. Similarly, lumacaftor has a positive impact on Y1092X, but not on N1303K. Our data also show that cells with only one copy of F508del-CFTR respond less to VX-809. Moreover, there is great variability in lumacaftor responses among F508del-homozygous cells from different donors. Compound C18 failed to rescue A561E-CFTR but not in F508del-CFTR, thus plausibly it has a different mechanism of action distinct from lumacaftor.
Conclusions: CF patients with A561E (and likely also those with Y1029X) can potentially benefit from lumacaftor. Moreover, the methodology used here exemplifies how ex vivo approaches may apply personalized therapies to CF and possibly other respiratory diseases.
Keywords: (ΔIeq-sc), equivalent short-circuit currents; CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; ENaC, epithelial Na+ channel; Fsk, forskolin; Gen, Genistein; HBE (cells), human bronchial epithelial cells; Innovative treatments; Mutation-specific therapies; Personalized medicine; Rare diseases; Rte, transepithelial resistance.; SEM, standard error of the mean; TEER, transepithelial electrical resistance; Vte, transepithelial voltage.
Figures


Comment in
-
Predicting CFTR activity with front-runner cystic fibrosis drugs.EBioMedicine. 2015 Jan 27;2(2):100-1. doi: 10.1016/j.ebiom.2015.01.013. eCollection 2015 Feb. EBioMedicine. 2015. PMID: 26137547 Free PMC article. No abstract available.
References
-
- Amaral M.D. CFTR and chaperones: processing and degradation. J. Mol. Neurosci. 2004;23(1–2):41–48. - PubMed
-
- Amaral M.D., Farinha C.M. Rescuing mutant CFTR: a multi-task approach to a better outcome in treating cystic fibrosis. Curr. Pharm. Des. 2013;19(19):3497–3508. - PubMed
-
- Bell S.C., De Boeck K., Amaral M.D. New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacol. Ther. 2015;145:19–34. - PubMed
-
- Bobadilla J.L., Macek M., Jr., Fine J.P., Farrell P.M. Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum. Mutat. 2002;19(6):575–606. - PubMed
-
- De Boeck K., Paskavitz J., Chen X., Higgins M. ivacaftor, a CFTR potentiator, in cystic fibrosis patients who have a non-G551D-CFTR gating mutation: phase 3, part 1 results. Pediatr. Pulmonol. 2013;48(36):292.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
