

Activation of Autophagic Flux against Xenoestrogen Bisphenol-A-induced Hippocampal Neurodegeneration via AMP kinase (AMPK)/Mammalian Target of Rapamycin (mTOR) Pathways
- PMID: 26139607
- PMCID: PMC4543672
- DOI: 10.1074/jbc.M115.648998
Activation of Autophagic Flux against Xenoestrogen Bisphenol-A-induced Hippocampal Neurodegeneration via AMP kinase (AMPK)/Mammalian Target of Rapamycin (mTOR) Pathways
Retraction in
-
Withdrawal: Activation of autophagic flux against xenoestrogen bisphenol-A-Induced hippocampal neurodegeneration via AMP kinase (AMPK)/mammalian target of rapamycin (mTOR) pathways.J Biol Chem. 2020 Feb 28;295(9):2889. doi: 10.1074/jbc.W120.012895. Epub 2020 Feb 28. J Biol Chem. 2020. PMID: 32111724 Free PMC article. No abstract available.
Abstract
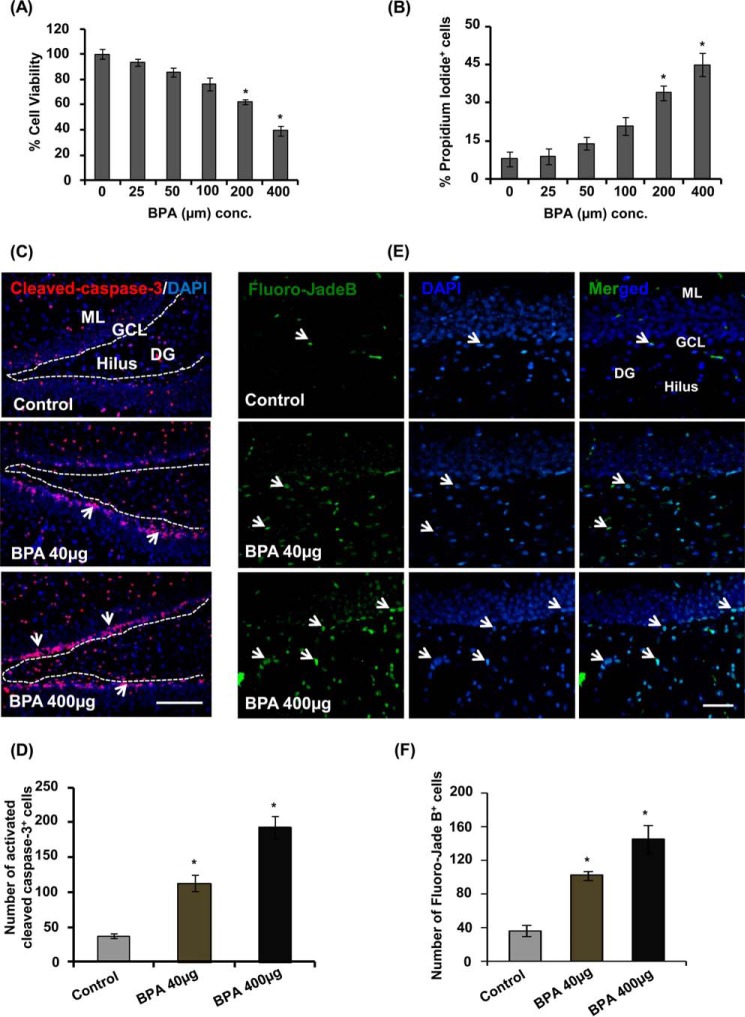
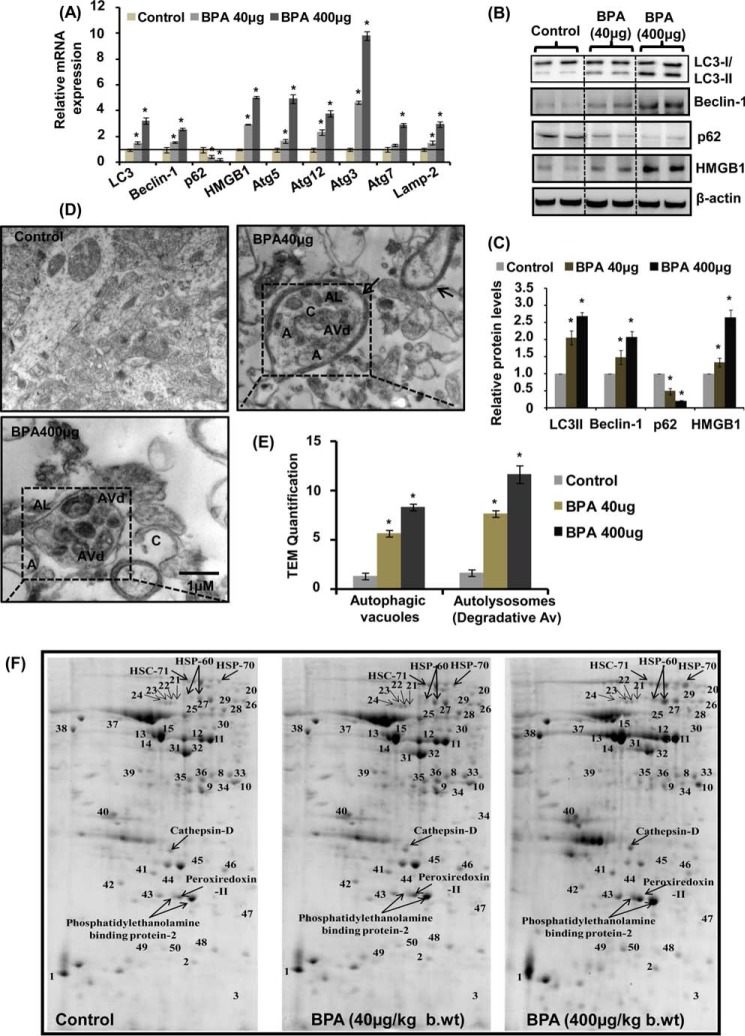
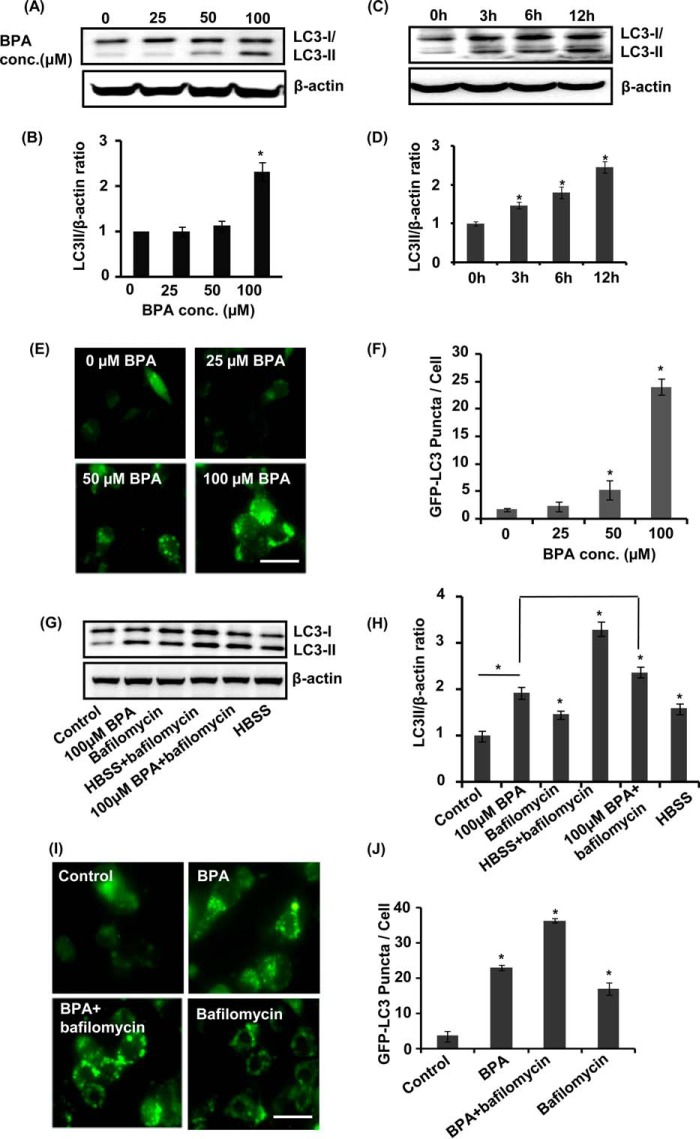
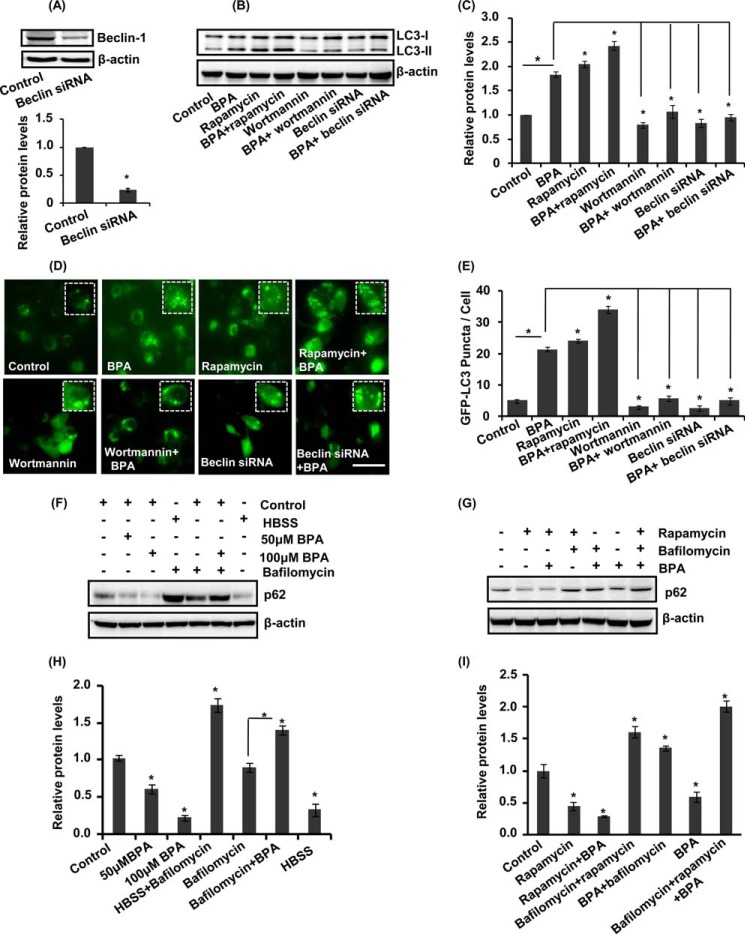
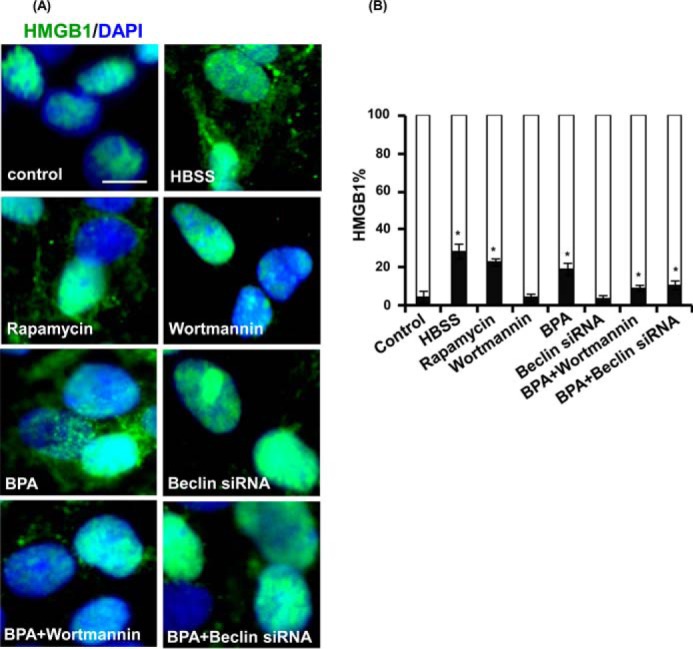
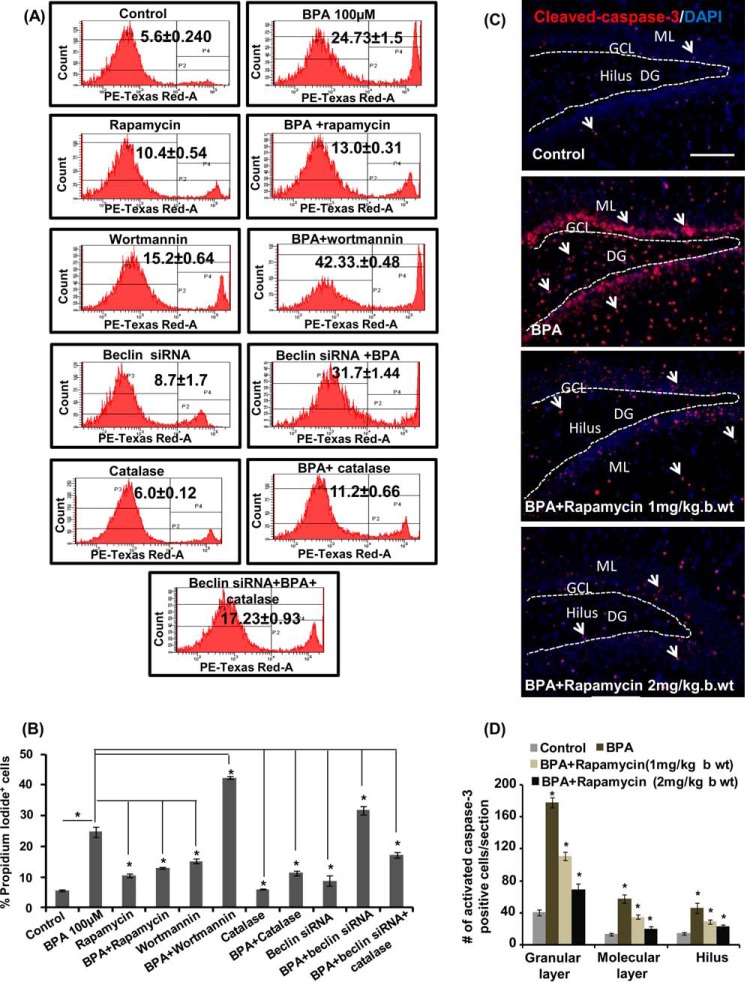
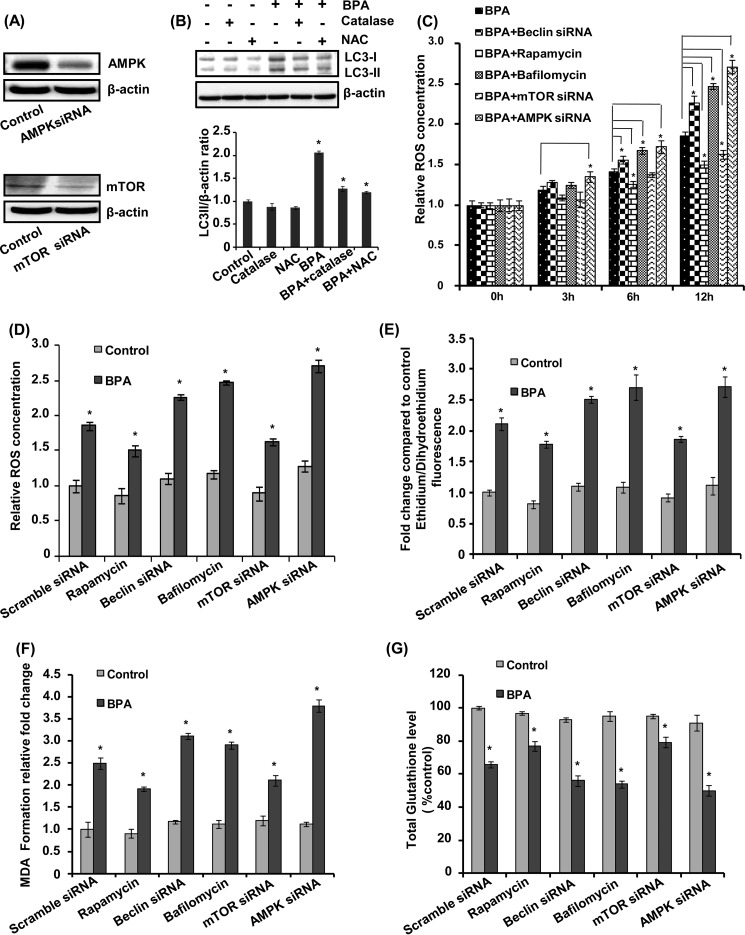
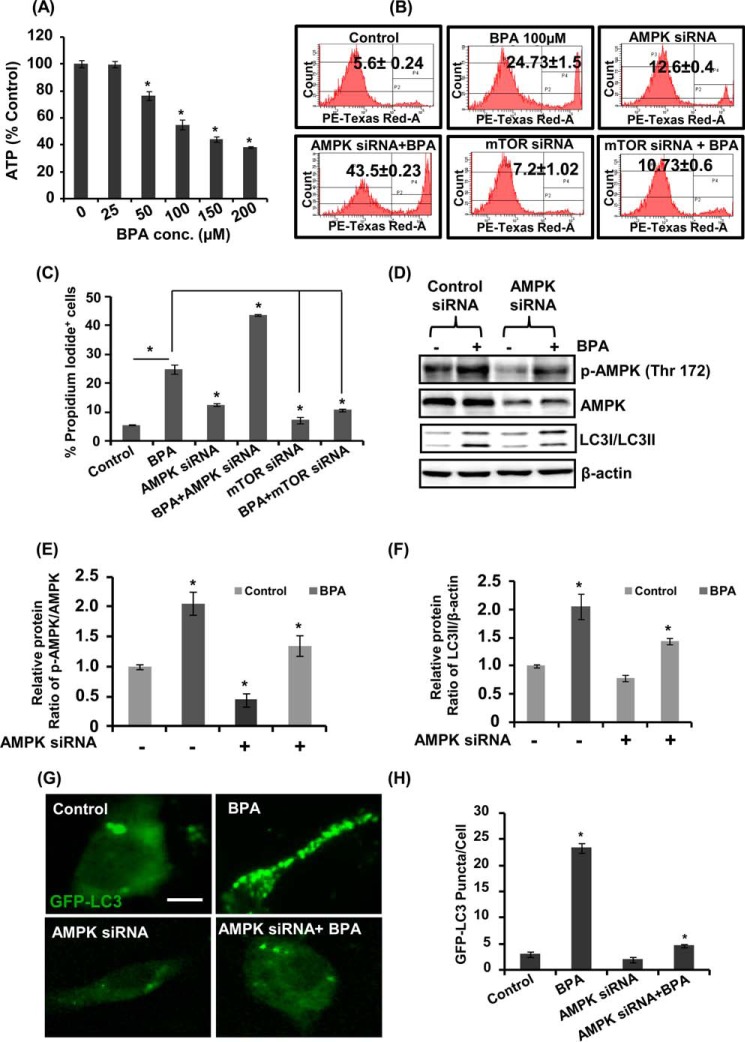
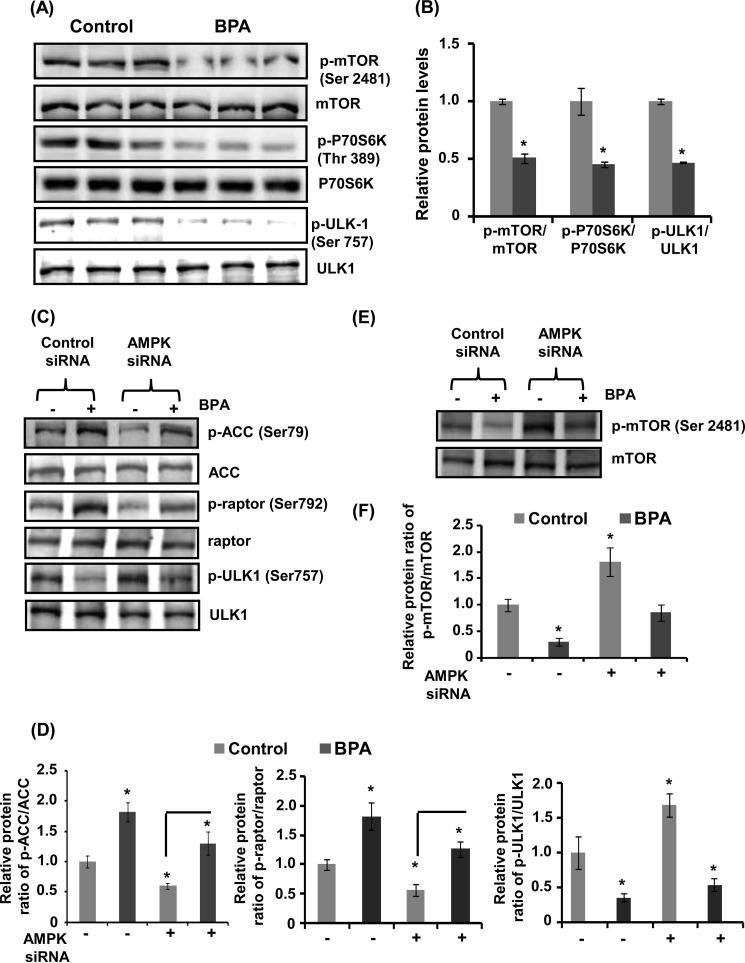
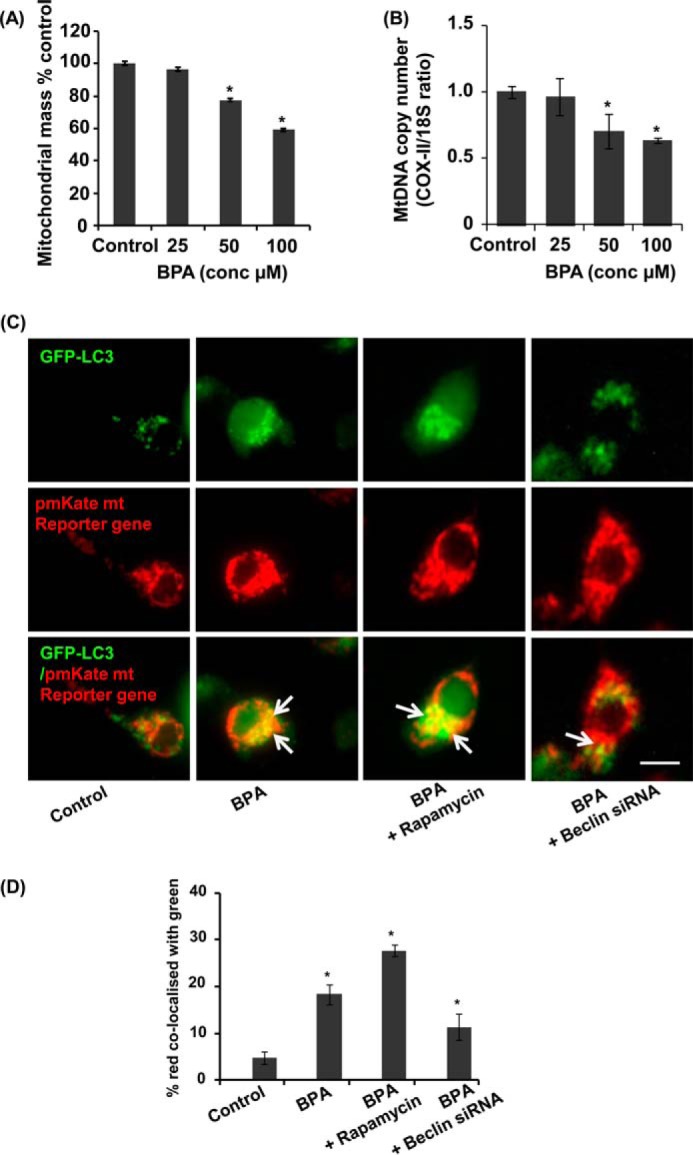
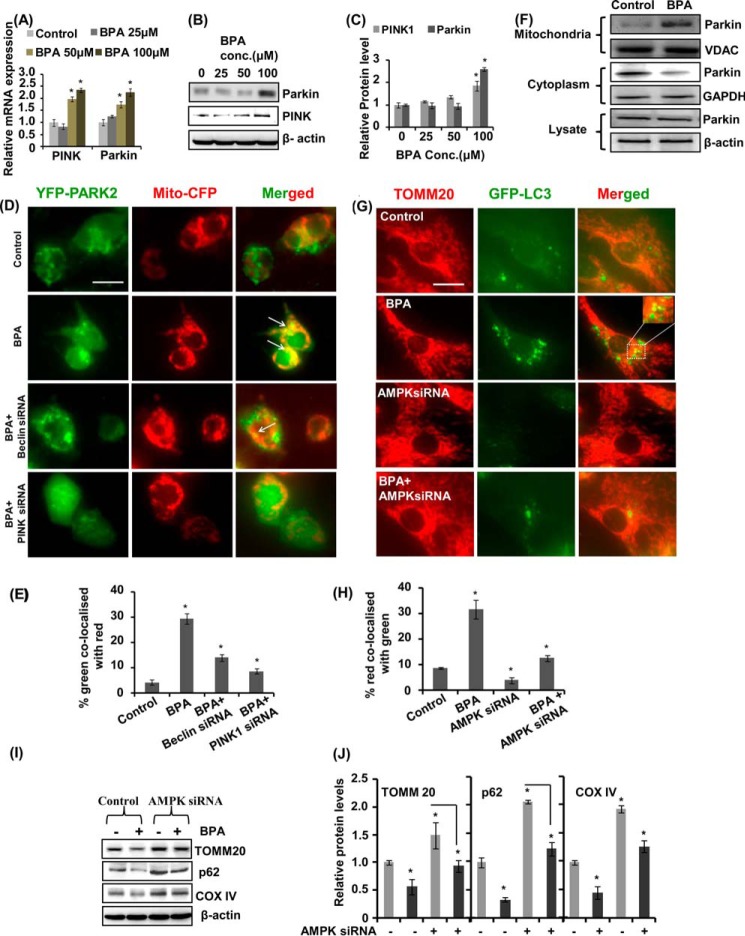
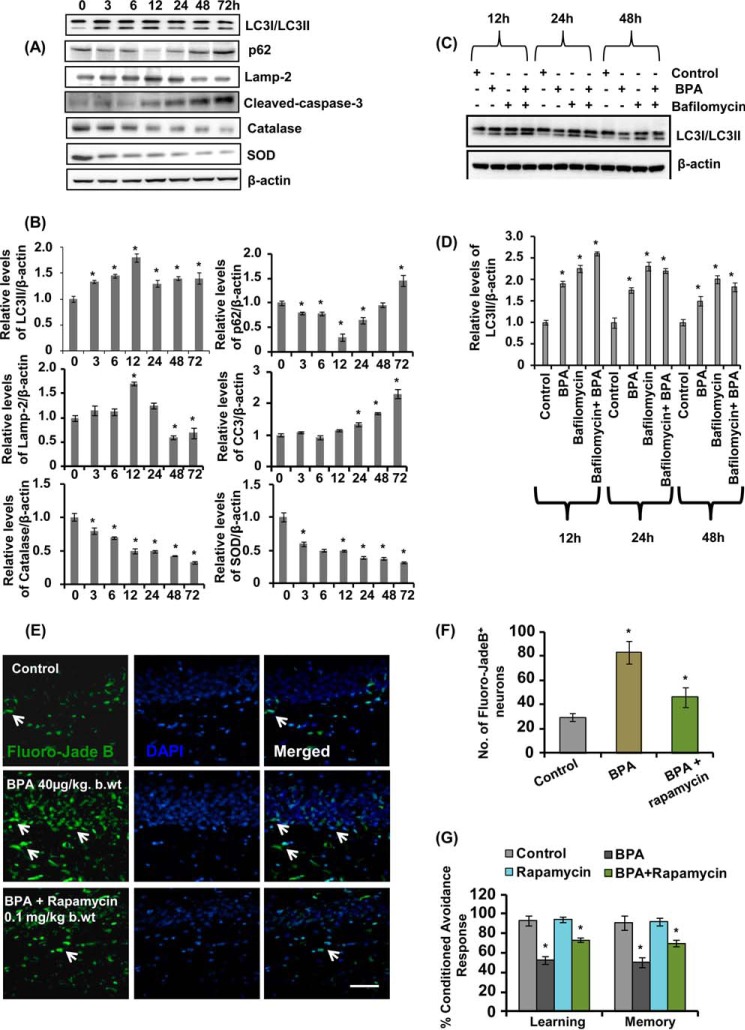
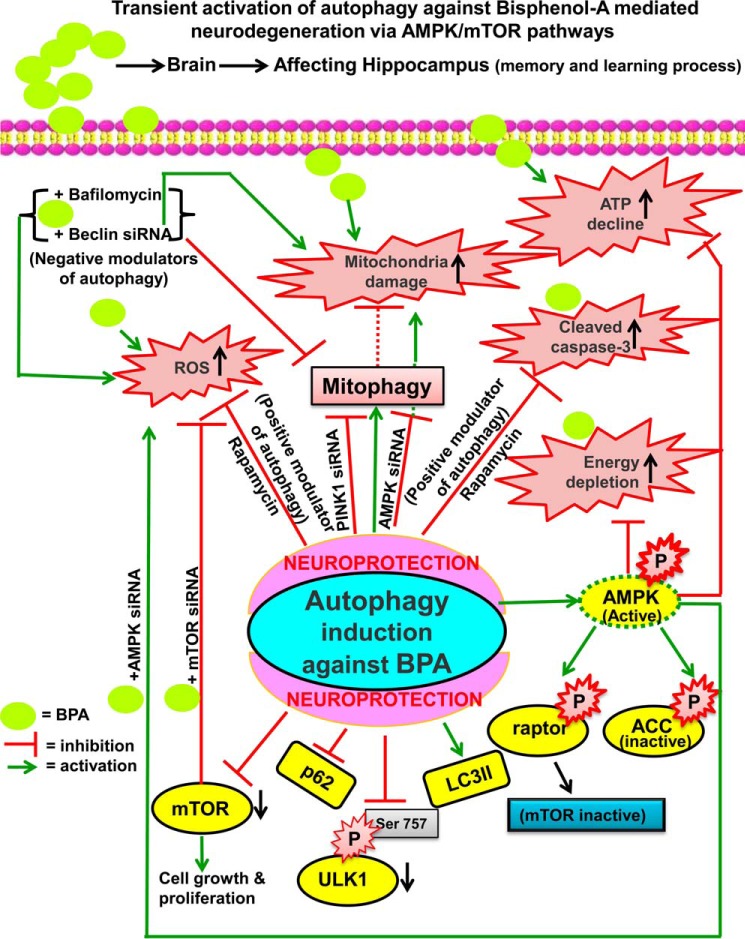
The human health hazards related to persisting use of bisphenol-A (BPA) are well documented. BPA-induced neurotoxicity occurs with the generation of oxidative stress, neurodegeneration, and cognitive dysfunctions. However, the cellular and molecular mechanism(s) of the effects of BPA on autophagy and association with oxidative stress and apoptosis are still elusive. We observed that BPA exposure during the early postnatal period enhanced the expression and the levels of autophagy genes/proteins. BPA treatment in the presence of bafilomycin A1 increased the levels of LC3-II and SQSTM1 and also potentiated GFP-LC3 puncta index in GFP-LC3-transfected hippocampal neural stem cell-derived neurons. BPA-induced generation of reactive oxygen species and apoptosis were mitigated by a pharmacological activator of autophagy (rapamycin). Pharmacological (wortmannin and bafilomycin A1) and genetic (beclin siRNA) inhibition of autophagy aggravated BPA neurotoxicity. Activation of autophagy against BPA resulted in intracellular energy sensor AMP kinase (AMPK) activation, increased phosphorylation of raptor and acetyl-CoA carboxylase, and decreased phosphorylation of ULK1 (Ser-757), and silencing of AMPK exacerbated BPA neurotoxicity. Conversely, BPA exposure down-regulated the mammalian target of rapamycin (mTOR) pathway by phosphorylation of raptor as a transient cell's compensatory mechanism to preserve cellular energy pool. Moreover, silencing of mTOR enhanced autophagy, which further alleviated BPA-induced reactive oxygen species generation and apoptosis. BPA-mediated neurotoxicity also resulted in mitochondrial loss, bioenergetic deficits, and increased PARKIN mitochondrial translocation, suggesting enhanced mitophagy. These results suggest implication of autophagy against BPA-mediated neurodegeneration through involvement of AMPK and mTOR pathways. Hence, autophagy, which arbitrates cell survival and demise during stress conditions, requires further assessment to be established as a biomarker of xenoestrogen exposure.
Keywords: hippocampus; neural stem cell (NSC); neurodegeneration; toxicology; xenobiotic.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Jain S., Kumar C. H., Suranagi U. D., Mediratta P. K. (2011) Protective effect of N-acetylcysteine on bisphenol A-induced cognitive dysfunction and oxidative stress in rats. Food Chem. Toxicol. 49, 1404–1409 - PubMed
-
- Itoh K., Yaoi T., Fushiki S. (2012) Bisphenol A, an endocrine-disrupting chemical, and brain development. Neuropathology 32, 447–457 - PubMed
-
- Ishido M., Masuo Y., Terasaki M., Morita M. (2011) Rat hyperactivity by bisphenol A, but not by its derivatives, 3-hydroxybisphenol A or bisphenol A 3,4-quinone. Toxicol. Lett. 206, 300–305 - PubMed
-
- Lee S., Kim Y. K., Shin T. Y., Kim S. H. (2013) Neurotoxic effects of bisphenol AF on calcium-induced ROS and MAPKs. Neurotox. Res. 23, 249–259 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
