

Nicotinic Acetylcholine Receptors Sensitize a MAPK-linked Toxicity Pathway on Prolonged Exposure to β-Amyloid
- PMID: 26139609
- PMCID: PMC4571869
- DOI: 10.1074/jbc.M114.634162
Nicotinic Acetylcholine Receptors Sensitize a MAPK-linked Toxicity Pathway on Prolonged Exposure to β-Amyloid
Abstract
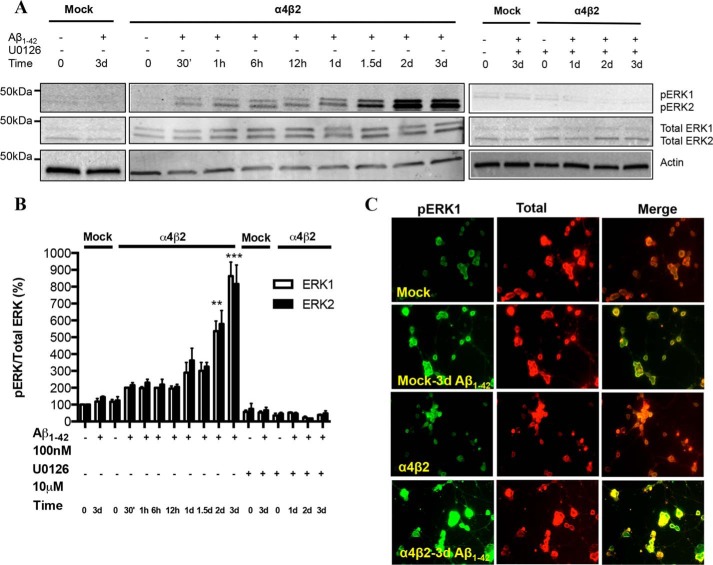
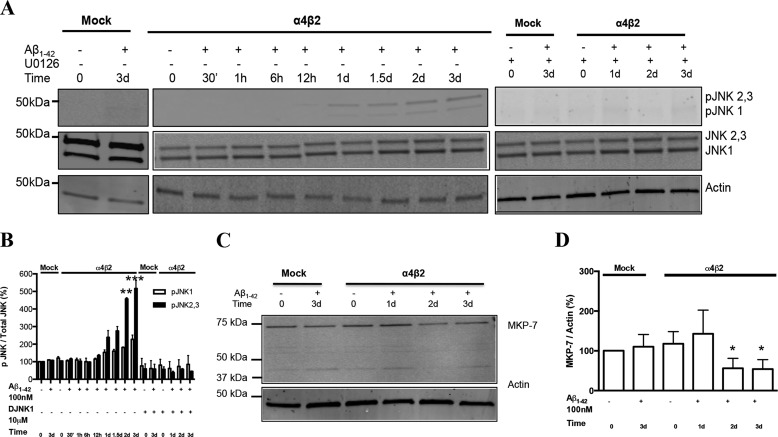
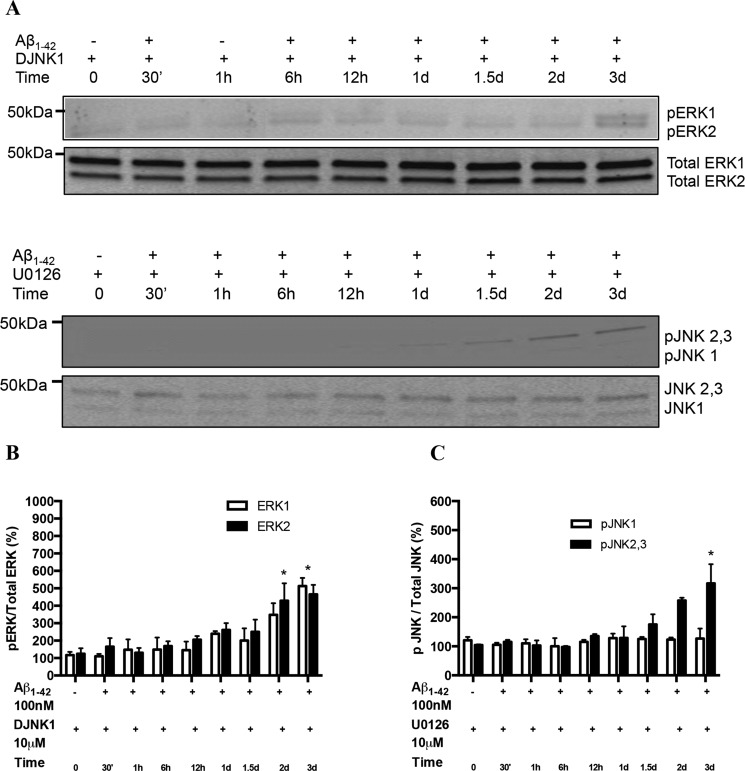
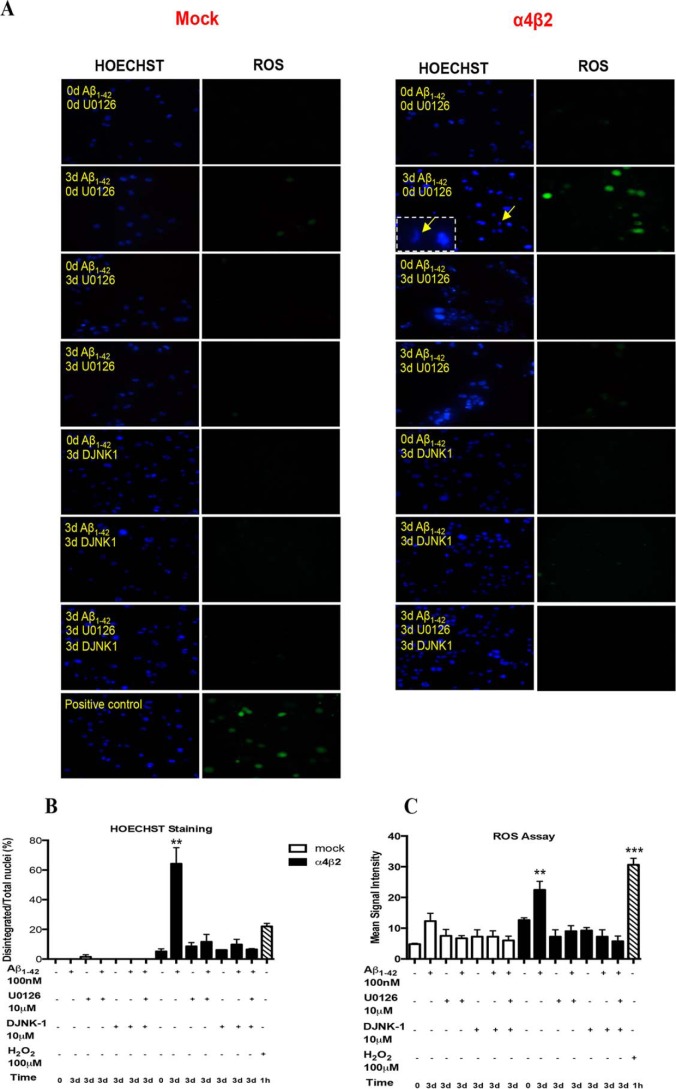
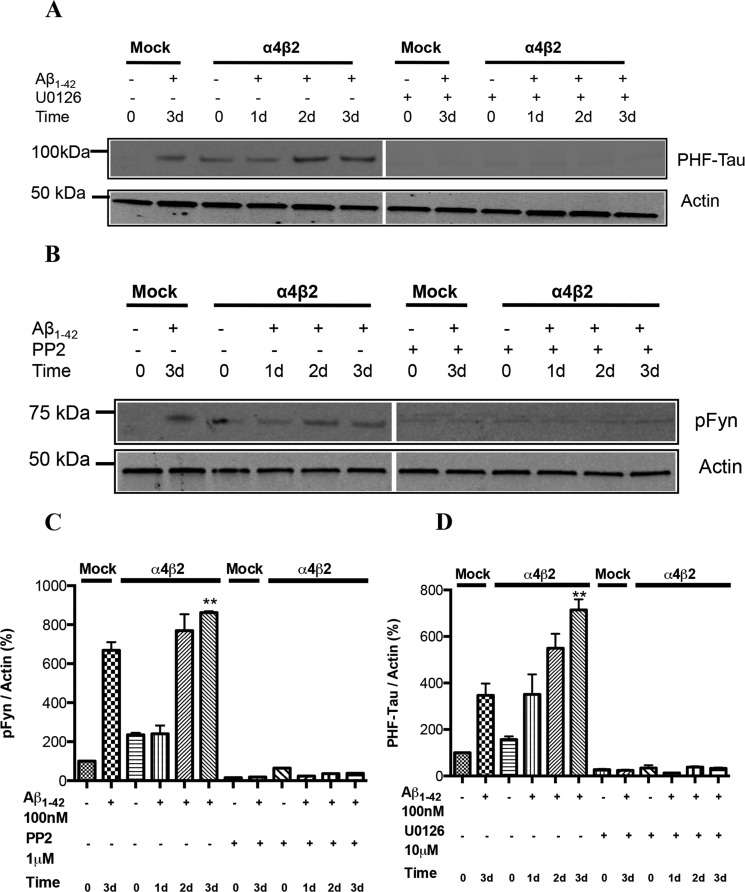
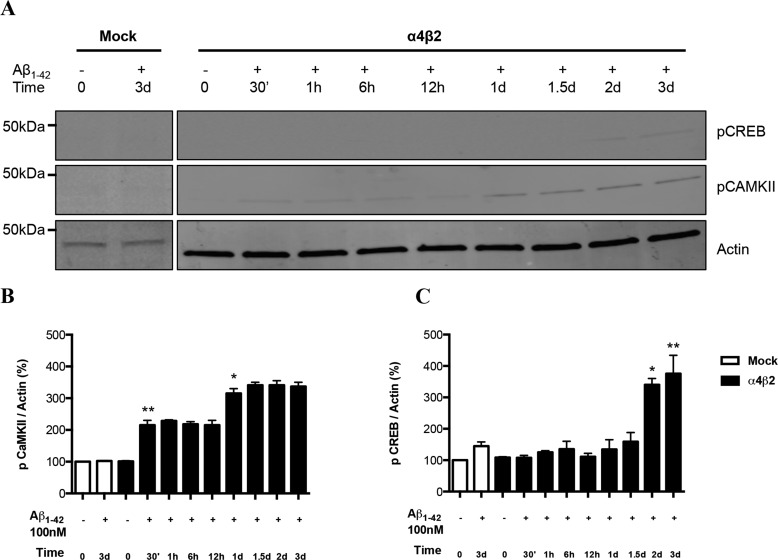
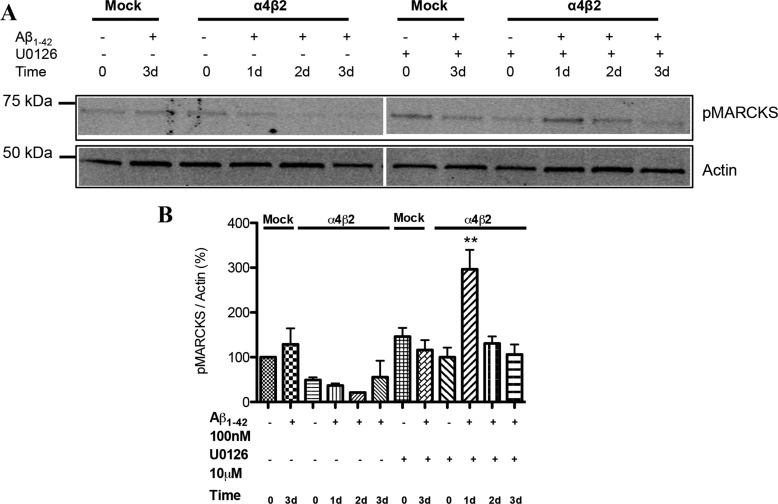
Among putative downstream synaptic targets of β-amyloid (Aβ) are signaling molecules involved in synaptic function, memory formation and cognition, such as the MAP kinases, MKPs, CaMKII, CREB, Fyn, and Tau. Here, we assessed the activation and interaction of signaling pathways upon prolonged exposure to Aβ in model nerve cells expressing nicotinic acetylcholine receptors (nAChRs). Our goal was to characterize the steps underlying sensitization of the nerve cells to neurotoxicity when Aβ-target receptors are present. Of particular focus was the connection of the activated signaling molecules to oxidative stress. Differentiated neuroblastoma cells expressing mouse α4β2-nAChRs were exposed to Aβ1-42 for intervals from 30 min to 3 days. The cells and cell-derived protein extracts were then probed for activation of signaling pathway molecules (ERK, JNK, CaMKII, CREB, MARCKS, Fyn, tau). Our results show substantial, progressive activation of ERK in response to nanomolar Aβ exposure, starting at the earliest time point. Increased ERK activation was followed by JNK activation as well as an increased expression of PHF-tau, paralleled by increased levels of reactive oxygen species (ROS). The impact of prolonged Aβ on the levels of pERK, pJNK, and ROS was attenuated by MEK-selective and JNK-selective inhibitors. In addition, the MEK inhibitor as well as a JNK inhibitor attenuated Aβ-induced nuclear fragmentation, which followed the changes in ROS levels. These results demonstrate that the presence of nAChRs sensitizes neurons to the neurotoxic action of Aβ through the timed activation of discrete intracellular signaling molecules, suggesting pathways involved in the early stages of Alzheimer disease.
Keywords: amyloid-beta (AB); c-Jun N-terminal kinase (JNK); extracellular-signal-regulated kinase (ERK); mitogen-activated protein kinase (MAPK); nicotinic acetylcholine receptors (nAChR).
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures

References
-
- Balleza-Tapia H., Peña F. (2009) Pharmacology of the intracellular pathways activated by Amyloid beta protein. Mini-Rev. Med. Chem. 9, 724–740 - PubMed
-
- Pei J. J., Braak E., Braak H., Grundke-Iqbal I., Iqbal K., Winblad B., Cowburn R. F. (2001) Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer's disease brains at different stages of neurofibrillary degeneration. J. Alzheimers Dis. 3, 41–48 - PubMed
-
- Ferrer I., Blanco R., Carmona M., Ribera R., Goutan E., Puig B., Rey M. J., Cardozo A., Viñals R., Ribalta T. (2001) Phosphorylated MAP kinase (ERK1, ERK2) expression is associated with early tau deposition in neurons and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer's disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 11, 144–158 - PMC - PubMed
-
- Dineley K. T., Westerman M., Bui D., Bell K., Ashe K. H., Sweatt J. D. (2001) Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J. Neurosci. 21, 4125–4133 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
