

A bioinformatic framework for immune repertoire diversity profiling enables detection of immunological status
- PMID: 26140055
- PMCID: PMC4489130
- DOI: 10.1186/s13073-015-0169-8
A bioinformatic framework for immune repertoire diversity profiling enables detection of immunological status
Abstract
Background: Lymphocyte receptor repertoires are continually shaped throughout the lifetime of an individual in response to environmental and pathogenic exposure. Thus, they may serve as a fingerprint of an individual's ongoing immunological status (e.g., healthy, infected, vaccinated), with far-reaching implications for immunodiagnostics applications. The advent of high-throughput immune repertoire sequencing now enables the interrogation of immune repertoire diversity in an unprecedented and quantitative manner. However, steadily increasing sequencing depth has revealed that immune repertoires vary greatly among individuals in their composition; correspondingly, it has been reported that there are few shared sequences indicative of immunological status ('public clones'). Disconcertingly, this means that the wealth of information gained from repertoire sequencing remains largely unused for determining the current status of immune responses, thereby hampering the implementation of immune-repertoire-based diagnostics.
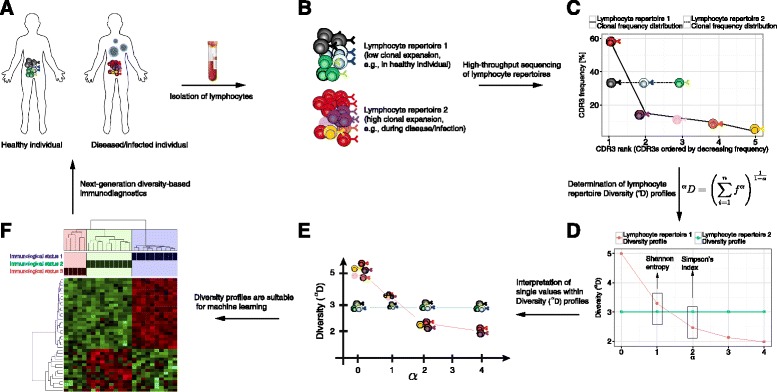
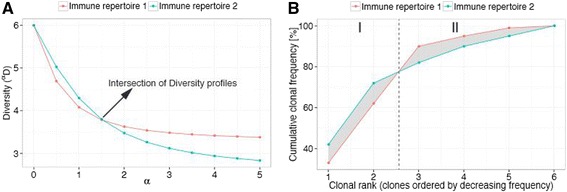
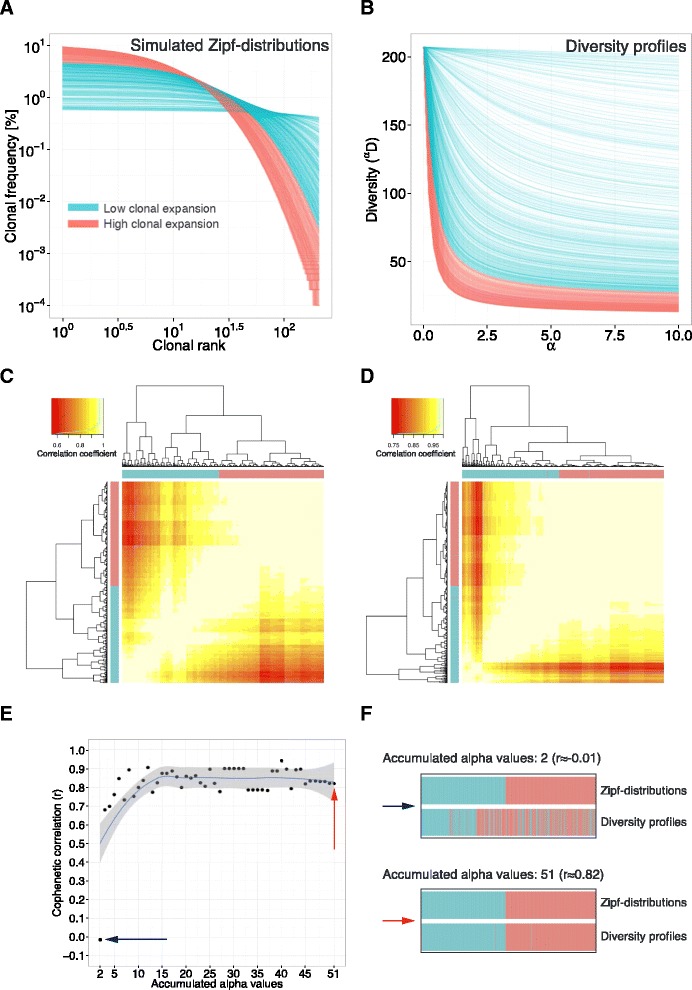
Methods: Here, we introduce a bioinformatics repertoire-profiling framework that possesses the advantage of capturing the diversity and distribution of entire immune repertoires, as opposed to singular public clones. The framework relies on Hill-based diversity profiles composed of a continuum of single diversity indices, which enable the quantification of the extent of immunological information contained in immune repertoires.
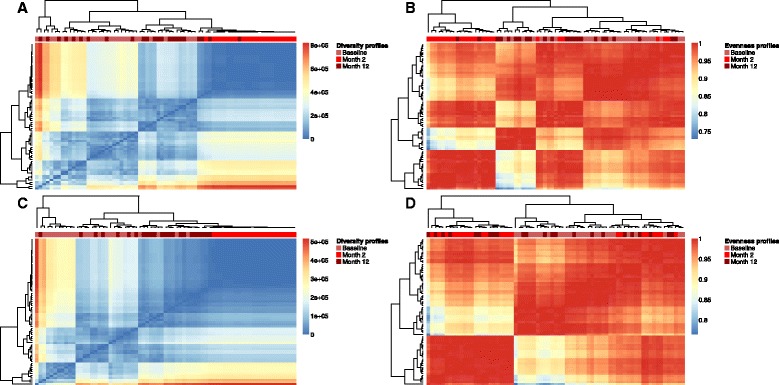
Results: We coupled diversity profiles with unsupervised (hierarchical clustering) and supervised (support vector machine and feature selection) machine learning approaches in order to correlate patients' immunological statuses with their B- and T-cell repertoire data. We could predict with high accuracy (greater than or equal to 80 %) a wide range of immunological statuses such as healthy, transplantation recipient, and lymphoid cancer, suggesting as a proof of principle that diversity profiling can recover a large amount of immunodiagnostic fingerprints from immune repertoire data. Our framework is highly scalable as it easily allowed for the analysis of 1000 simulated immune repertoires; this exceeds the size of published immune repertoire datasets by one to two orders of magnitude.
Conclusions: Our framework offers the possibility to advance immune-repertoire-based fingerprinting, which may in the future enable a systems immunogenomics approach for vaccine profiling and the accurate and early detection of disease and infection.
Figures
References
-
- Abbas AK, Lichtman A. Cellular and molecular immunology. 5. Philadelphia: Saunders; 2005.
LinkOut - more resources
Full Text Sources
Other Literature Sources
