

Disentangling the Effects of Colocalizing Genomic Annotations to Functionally Prioritize Non-coding Variants within Complex-Trait Loci
- PMID: 26140449
- PMCID: PMC4572568
- DOI: 10.1016/j.ajhg.2015.05.016
Disentangling the Effects of Colocalizing Genomic Annotations to Functionally Prioritize Non-coding Variants within Complex-Trait Loci
Abstract
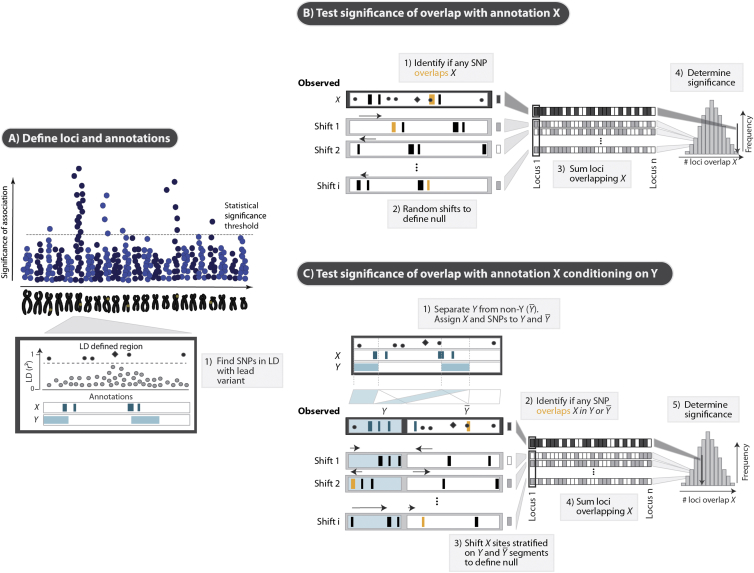
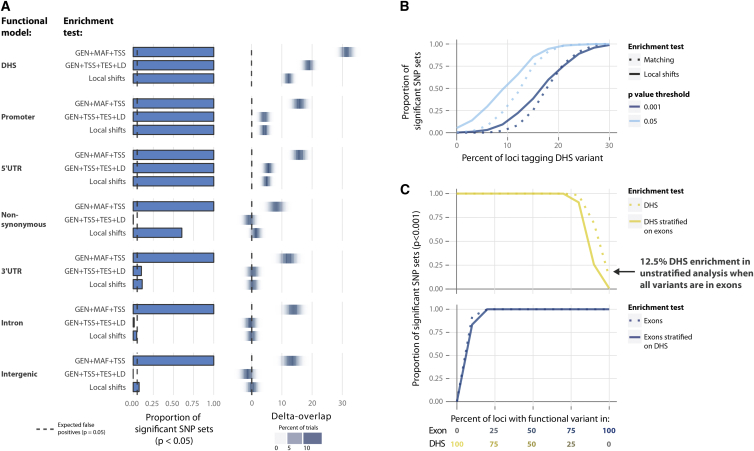
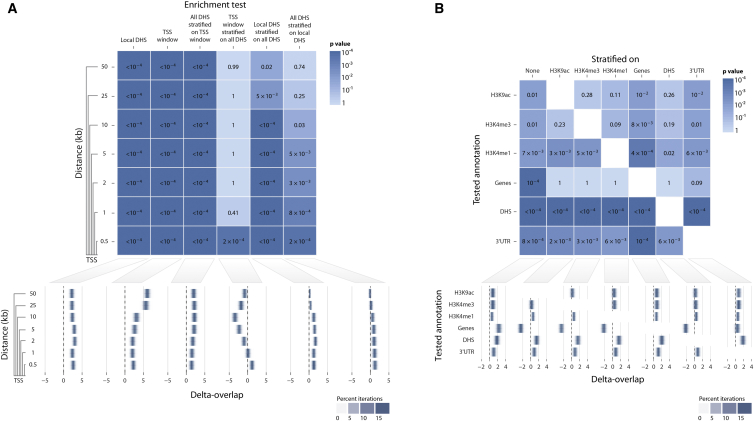
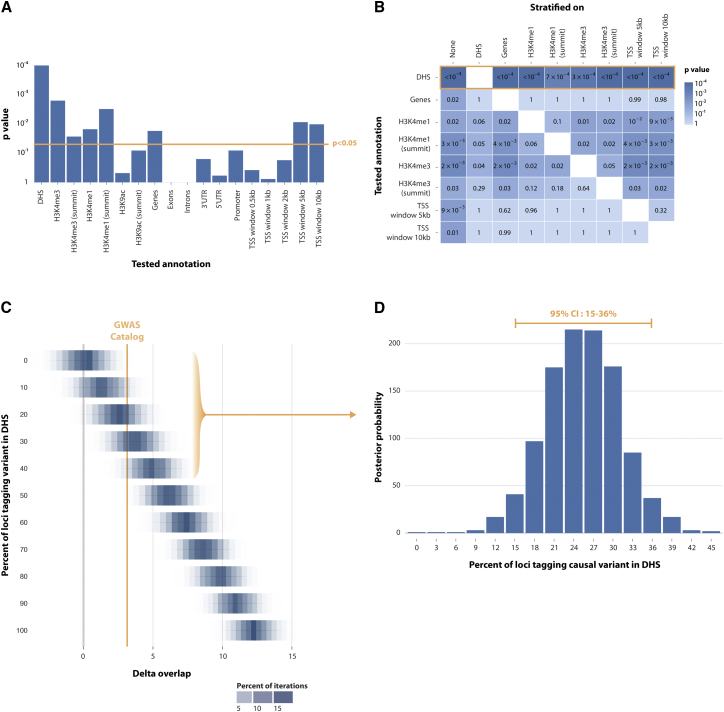
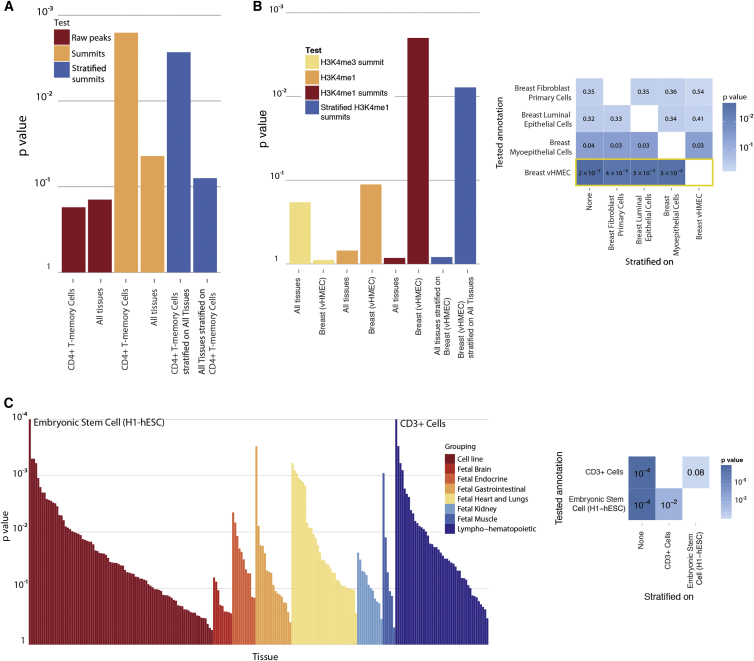
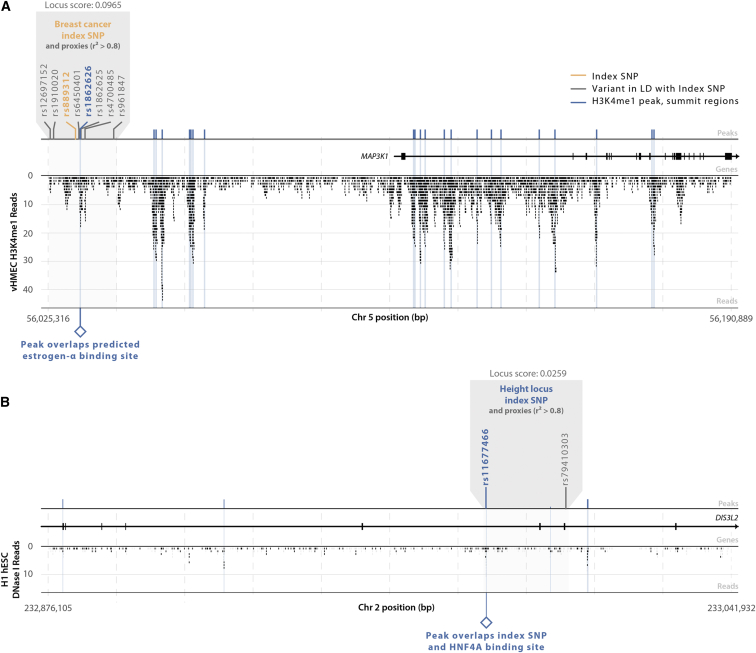
Identifying genomic annotations that differentiate causal from trait-associated variants is essential to fine mapping disease loci. Although many studies have identified non-coding functional annotations that overlap disease-associated variants, these annotations often colocalize, complicating the ability to use these annotations for fine mapping causal variation. We developed a statistical approach (Genomic Annotation Shifter [GoShifter]) to assess whether enriched annotations are able to prioritize causal variation. GoShifter defines the null distribution of an annotation overlapping an allele by locally shifting annotations; this approach is less sensitive to biases arising from local genomic structure than commonly used enrichment methods that depend on SNP matching. Local shifting also allows GoShifter to identify independent causal effects from colocalizing annotations. Using GoShifter, we confirmed that variants in expression quantitative trail loci drive gene-expression changes though DNase-I hypersensitive sites (DHSs) near transcription start sites and independently through 3' UTR regulation. We also showed that (1) 15%-36% of trait-associated loci map to DHSs independently of other annotations; (2) loci associated with breast cancer and rheumatoid arthritis harbor potentially causal variants near the summits of histone marks rather than full peak bodies; (3) variants associated with height are highly enriched in embryonic stem cell DHSs; and (4) we can effectively prioritize causal variation at specific loci.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Allele-specific binding variants causing ChIP-seq peak height of histone modification are not enriched in expression QTL annotations.Genet Sel Evol. 2024 Jun 27;56(1):50. doi: 10.1186/s12711-024-00916-4. Genet Sel Evol. 2024. PMID: 38937662 Free PMC article.
-
Incorporating Functional Annotations for Fine-Mapping Causal Variants in a Bayesian Framework Using Summary Statistics.Genetics. 2016 Nov;204(3):933-958. doi: 10.1534/genetics.116.188953. Epub 2016 Sep 21. Genetics. 2016. PMID: 27655946 Free PMC article.
-
The genomic signature of trait-associated variants.BMC Genomics. 2013 Feb 18;14:108. doi: 10.1186/1471-2164-14-108. BMC Genomics. 2013. PMID: 23418889 Free PMC article.
-
Expression Quantitative Trait Loci Information Improves Predictive Modeling of Disease Relevance of Non-Coding Genetic Variation.PLoS One. 2015 Oct 16;10(10):e0140758. doi: 10.1371/journal.pone.0140758. eCollection 2015. PLoS One. 2015. PMID: 26474488 Free PMC article. Review.
-
Strengthening Causal Inference for Complex Disease Using Molecular Quantitative Trait Loci.Trends Mol Med. 2020 Feb;26(2):232-241. doi: 10.1016/j.molmed.2019.10.004. Epub 2019 Nov 9. Trends Mol Med. 2020. PMID: 31718940 Review.
Cited by
-
Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types.Nat Genet. 2017 Apr;49(4):600-605. doi: 10.1038/ng.3795. Epub 2017 Feb 20. Nat Genet. 2017. PMID: 28218759 Free PMC article.
-
Combining artificial intelligence: deep learning with Hi-C data to predict the functional effects of non-coding variants.Bioinformatics. 2021 Jun 16;37(10):1339-1344. doi: 10.1093/bioinformatics/btaa970. Bioinformatics. 2021. PMID: 33196774 Free PMC article.
-
Genetic predisposition to mosaic Y chromosome loss in blood.Nature. 2019 Nov;575(7784):652-657. doi: 10.1038/s41586-019-1765-3. Epub 2019 Nov 20. Nature. 2019. PMID: 31748747 Free PMC article.
-
Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes.Nat Genet. 2021 Jul;53(7):962-971. doi: 10.1038/s41588-021-00880-5. Epub 2021 Jun 14. Nat Genet. 2021. PMID: 34127860 Free PMC article.
-
Tropomyosin 1 genetically constrains in vitro hematopoiesis.BMC Biol. 2020 May 14;18(1):52. doi: 10.1186/s12915-020-00783-7. BMC Biol. 2020. PMID: 32408895 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
