

Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications
- PMID: 26142467
- PMCID: PMC7959410
- DOI: 10.1093/eurheartj/ehv305
Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications
Abstract
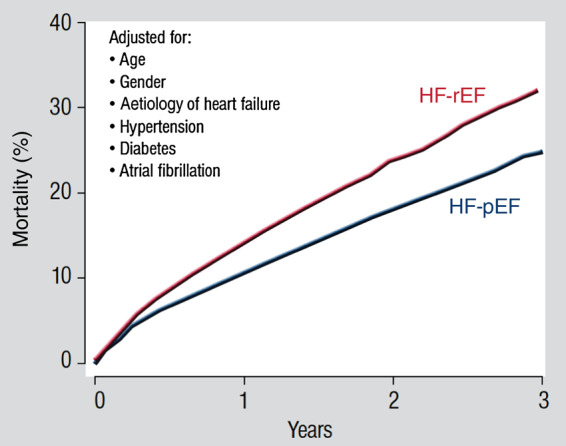
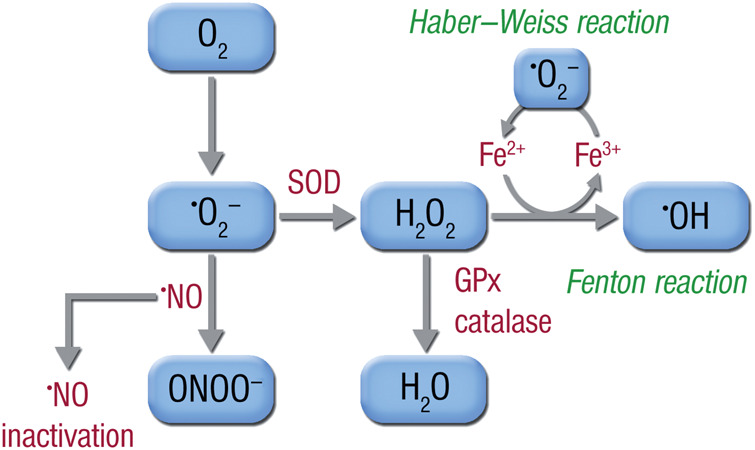
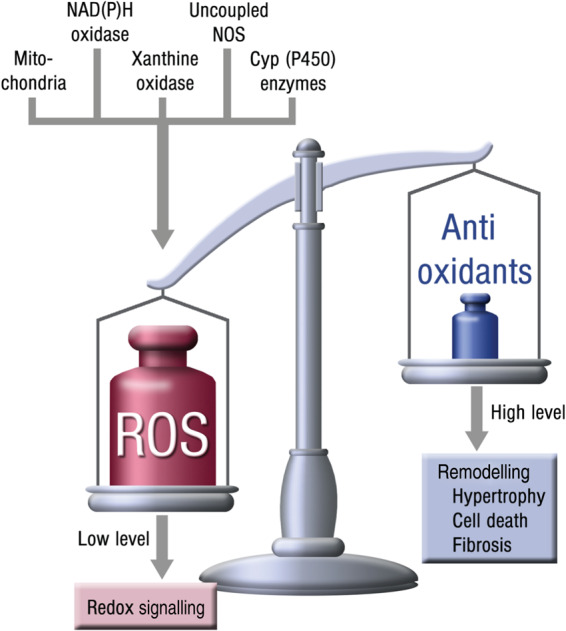
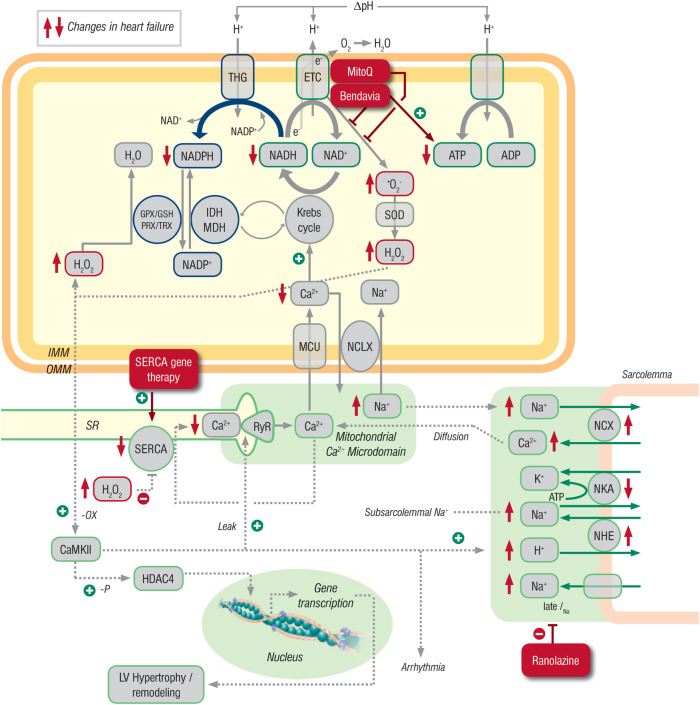
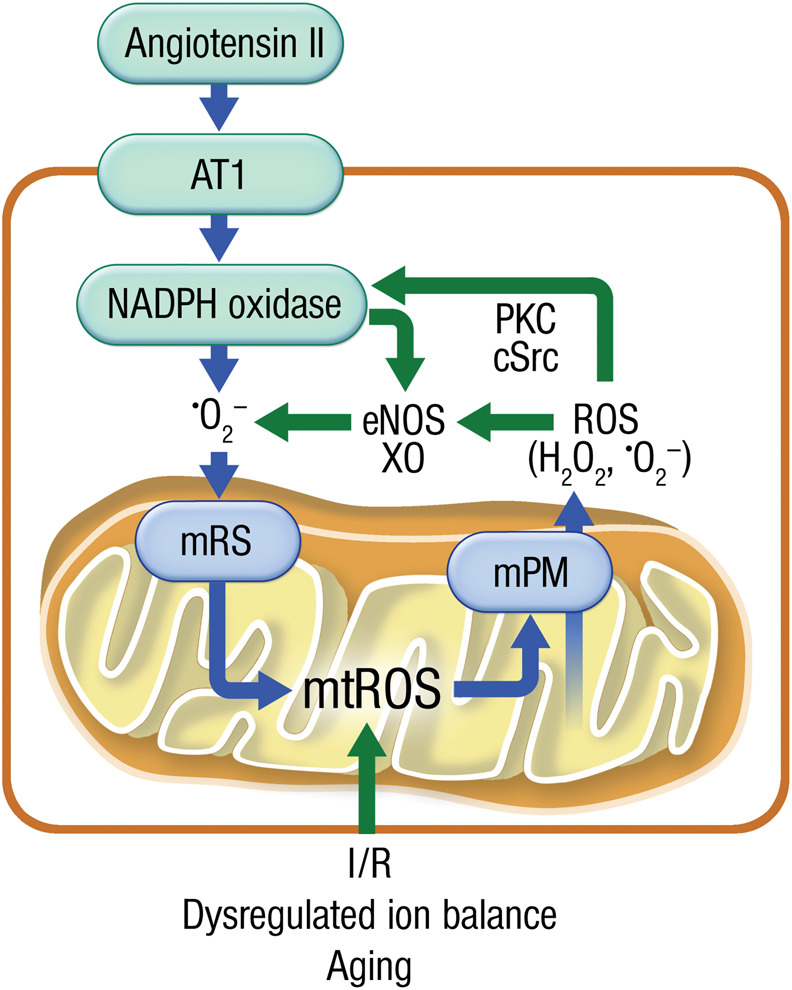
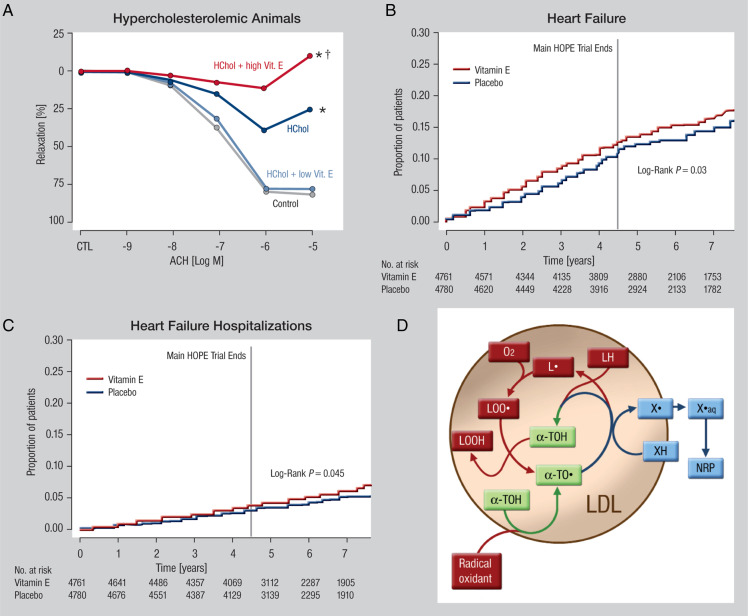
Systolic and diastolic myocardial dysfunction has been demonstrated to be associated with an activation of the circulating and local renin-angiotensin-aldosterone system (RAAS), and with a subsequent inappropriately increased production of reactive oxygen species (ROS). While, at low concentrations, ROS modulate important physiological functions through changes in cellular signalling and gene expression, overproduction of ROS may adversely alter cardiac mechanics, leading to further worsening of systolic and diastolic function. In addition, vascular endothelial dysfunction due to uncoupling of the nitric oxide synthase, activation of vascular and phagocytic membrane oxidases or mitochondrial oxidative stress may lead to increased vascular stiffness, further compromising cardiac performance in afterload-dependent hearts. In the present review, we address the potential role of ROS in the pathophysiology of myocardial and vascular dysfunction in heart failure (HF) and their therapeutic targeting. We discuss possible mechanisms underlying the failure of antioxidant vitamins in improving patients' prognosis, the impact of angiotensin-converting enzyme inhibitors or AT1 receptor blockers on oxidative stress, and the mechanism of the benefit of combination of hydralazine/isosorbide dinitrate. Further, we provide evidence supporting the existence of differences in the pathophysiology of HF with preserved vs. reduced ejection fraction and whether targeting mitochondrial ROS might be a particularly interesting therapeutic option for patients with preserved ejection fraction.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2015. For permissions please email: journals.permissions@oup.com.
Figures
References
-
- Cook C Cole G Asaria P Jabbour R Francis DP. The annual global economic burden of heart failure. Int J Cardiol 2014;171:368–376. - PubMed
-
- Meta-analysis Global Group in Chronic Heart Failure. The survival of patients with heart failure with preserved or reduced left ventricular ejection fraction: an individual patient data meta-analysis. Eur Heart J 2012;33:1750–1757. - PubMed
-
- Weber M Lauer N Mulsch A Kojda G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med 2001;31:1360–1367. - PubMed
-
- Forstermann U Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 2006;113:1708–1714. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
