

doi: 10.1038/nature14663.
Epub 2015 Jul 6.
Universal allosteric mechanism for Gα activation by GPCRs
Affiliations
- PMID: 26147082
- PMCID: PMC4866443
- DOI: 10.1038/nature14663
Item in Clipboard
Universal allosteric mechanism for Gα activation by GPCRs
Nature.
.
Abstract
G protein-coupled receptors (GPCRs) allosterically activate heterotrimeric G proteins and trigger GDP release. Given that there are ∼800 human GPCRs and 16 different Gα genes, this raises the question of whether a universal allosteric mechanism governs Gα activation. Here we show that different GPCRs interact with and activate Gα proteins through a highly conserved mechanism. Comparison of Gα with the small G protein Ras reveals how the evolution of short segments that undergo disorder-to-order transitions can decouple regions important for allosteric activation from receptor binding specificity. This might explain how the GPCR-Gα system diversified rapidly, while conserving the allosteric activation mechanism.
Figures
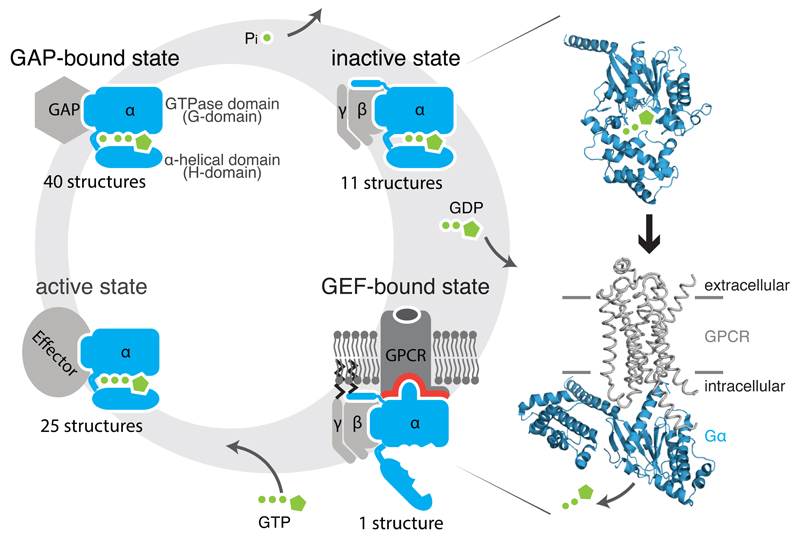
Heterotrimeric G proteins (1) release GDP upon binding to a GDP Exchange Factor (GEF), which are G protein-coupled receptors, (2) bind GTP and recruit downstream effectors, and (3) hydrolyze GTP promoted by GTPase activating protein (GAP), leading to (4) the inactive, GDP-bound state. Structures of the Gα subunit (blue) bound to GDP (1got; inactive state; top) and bound to the β2-AR (grey) (3sn6; active state; bottom) are shown.
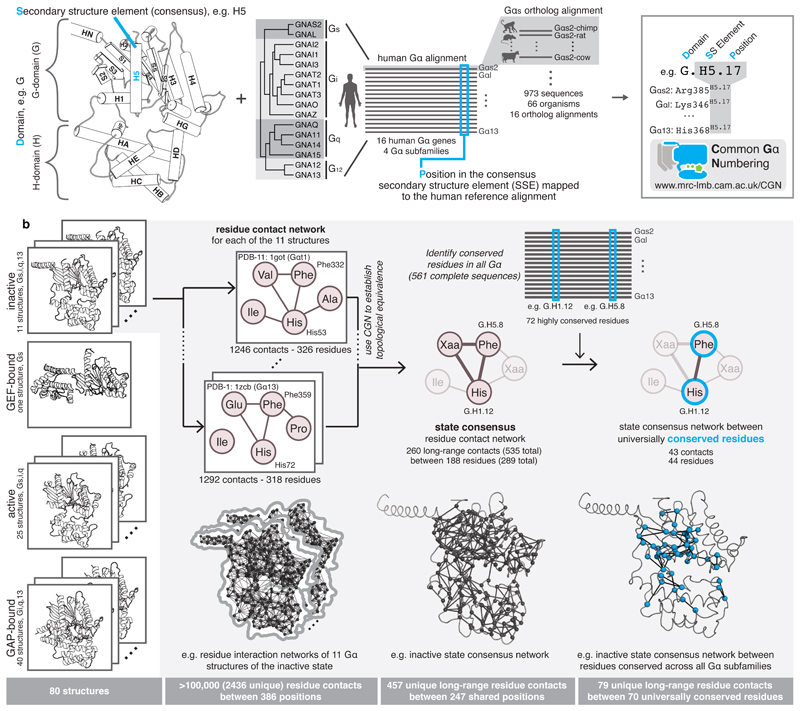
a, Every position in a Gα is denoted by its domain (D), the consensus secondary structure element where it is present (S), and position (P) within the consensus SSE. Names of SSEs are shown in the cartoon, loops are named with lower case of the flanking SSEs (e.g. h1ha; see Extended Figure 1 for all SSEs). An alignment of all 973 Gα proteins belonging to all the 4 subfamilies from 66 species allowed the identification of equivalent positions, P. An online webserver allows mapping of any Gα sequence or structure to the CGN system. b, Computation of consensus residue contact networks between conserved residues for different Gα signaling states. See Methods, Supplementary Note and Supplementary Data.
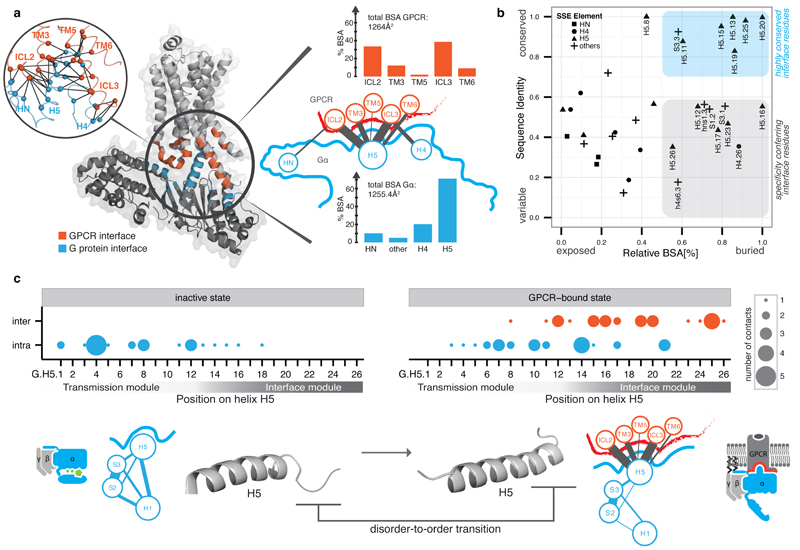
a, Inter Gα–GPCR residue contact network (inset) and buried surface area (BSA) analysis of the heterotrimeric Gαs–β2AR structure (3sn6). The line width between the nodes (SSE) denotes the number of consensus residue contacts. GPCR positions are denoted by extending the BW-numbering system (note parts of ICL3 become extended TM5). b, Scatterplot of Gα sequence conservation and normalized BSA highlights the conserved and variable interface residues. c, Consensus contact rewiring between the inactive and the GPCR-bound state by H5 residues. Positions mediating intra G protein contacts (blue) and receptor-mediated contacts (red) are shown. The circle size represents the number of contacts. H5 can be divided into transmission and interface module. The disorder-to-order transition of the H5 C-terminal region upon receptor binding and SSE contact rewiring are shown.
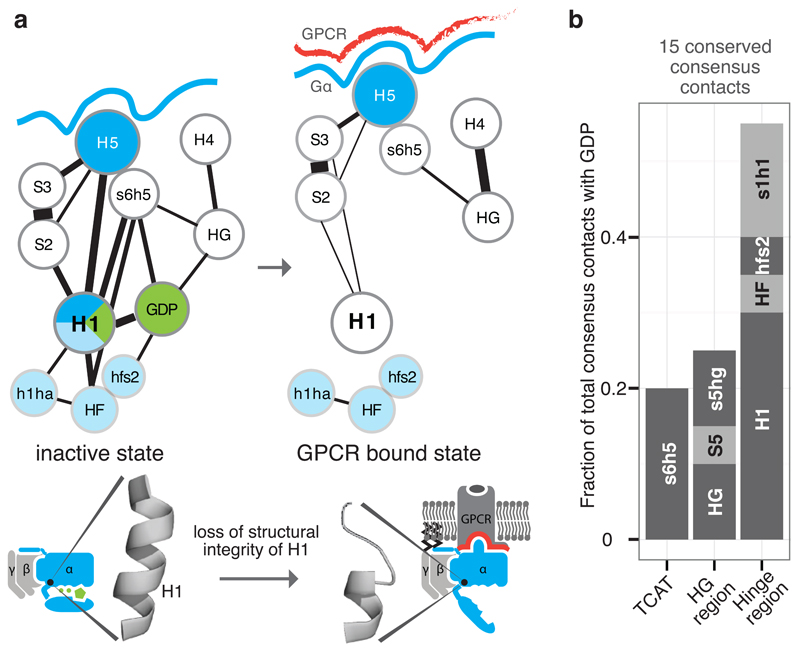
a, Consensus SSE contacts involving H1. The line width between the nodes (SSE) denotes the number of consensus residue contacts. Upon receptor binding, H5 is displaced and crucial contacts between H1 and H5 are lost. This might explain the increased flexibility of H1 in the GPCR-bound state, which results in the loss of GDP contacts and the H-domain hinge region (formed by H1, h1ha, and HF; light blue). b, The extent of GDP consensus contacts mediated by the different SSEs. See Extended Figure 4.

a, Disease and engineered mutations can be rationalized by the model of Gα activation in different Gα subfamilies. b, Comparison of the stability of Gαβγ-GDP (∆Tm; °C) and the Gαβγ-GPCR complex (∆ relative complex stability; %) by Gαi1 alanine mutagenesis of every position in H1 and H5. *Mutant FH5.8A is not stable in the receptor-free state but can still form the complex with the receptor.
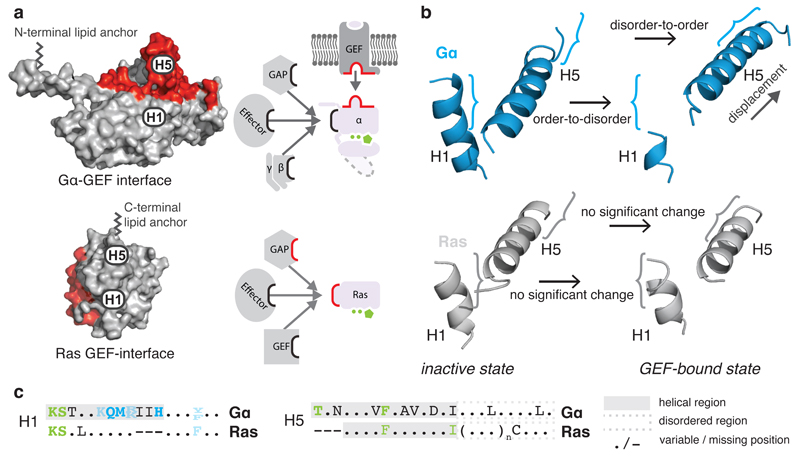
a, GEF interaction surfaces (red) for Gα (3sn6) and the small G protein HRas (1bkd) in a superimposed orientation. b, H1 and H5 of the inactive (1got, 4q21) and GEF-bound state (3sn6, 1bkd) for Gα (blue) and HRas (grey). c, Consensus sequences of equivalent residues of H5 and H1 in Gα and Ras. The helical region is highlighted in a grey background and the H5 disordered region is shown in a dashed grey box. See Extended Figure 6.

a, Reference alignment of all canonical human Gα paralogs. The domain (D), consensus secondary structure (S) and position in the SSE of the human reference alignment (P) are shown on top of the alignment. b, Reference table of the definitions of SSEs used in the CGN nomenclature.
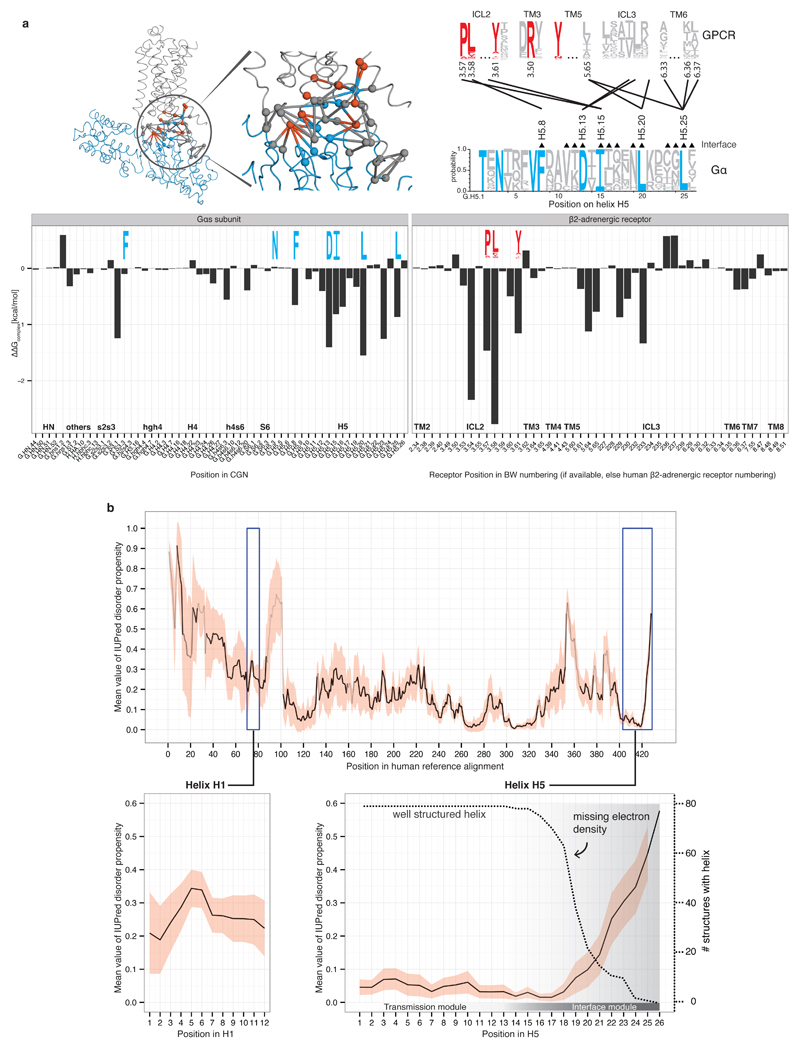
a, Energy contribution of single interface residues to the Gαs-β2AR complex calculated with FoldX (T = 298K, pH = 7.0, ion strength = 0.05M). Conserved Gα residues (blue sequence logo) that were identified to form receptor-Gα inter-protein contacts with conserved GPCR residues (red sequence logo) are shown. The contact network between residues of the β2AR and Gαs is shown (red, conserved receptor residue; blue, conserved Gα residue; grey, variable residues; spheres represent Cα positions and links represent non-covalent contact. b, Consensus disorder plot for all Gα proteins. The mean value of the disorder propensity of all full-length Gα sequences (561 sequences) from all 16 Gα types is shown as a black line, the standard deviation at each position is shown as light red ribbon. The color tone of the line indicates the number of gaps at an aligned position (black=no gaps). The left inset shows the disorder propensity of H1. The right inset highlights that H5 is highly structured in its N-terminus, and has increased disorder propensity towards the C-terminus, which is in agreement with the missing electron density in the 79 structures.
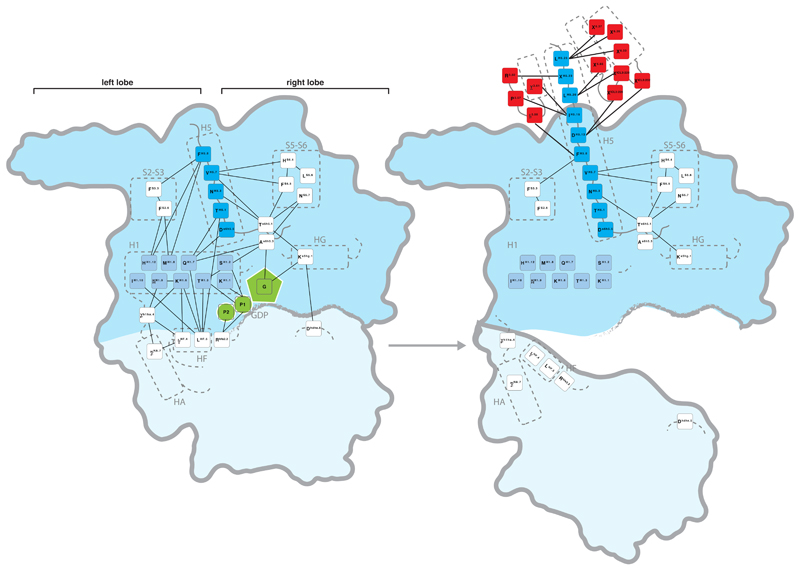
CGN numbers and sequence logo for consensus contacts within Gα in the inactive state (left) and GPCR-bound state (right) are shown. Receptor residues are shown in red, H5 residues in dark blue, H1 residues in light blue and GDP in green. The domains are highlighted with a blue background (G-domain darker blue, H-domain light blue).
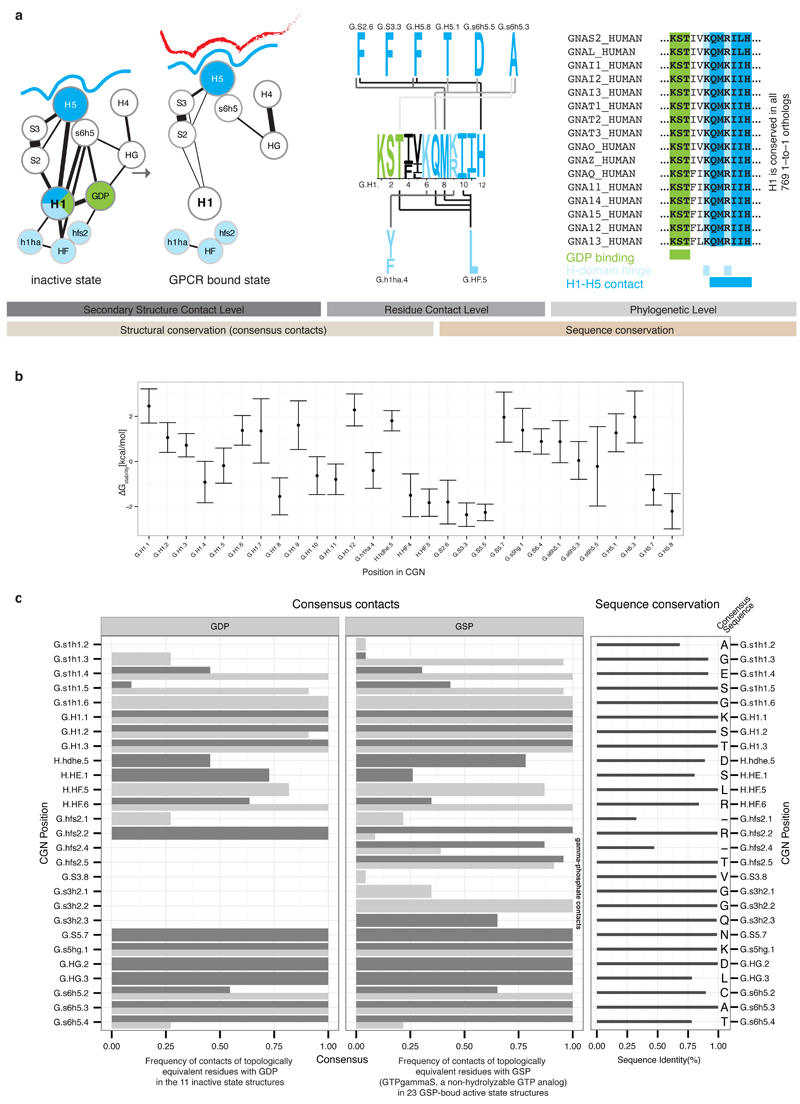
a, This Figure expands Figure 4 from the main text to provide residue-level details of the role of Helix H1. Residues forming contacts with H5 are shown in blue, with the H-domain in light blue and with GDP in green. Non-covalent consensus contacts between universally conserved residues at the SSE level (left) and per residue-level (center). Lines denote non-covalent contacts between residues. The degree of conservation is shown as sequence logo. Residues are numbered according to the CGN. Helix H1 is almost 100% conserved across all 16 Gα types and forms three structural motifs for interactions with H5, the H-domain and GDP (right). b, Average per residue energy contribution to Gα protein stability as calculated from 79 structures from all four Gα families of the non-receptor bound signaling states using FoldX (T = 298K, pH = 7.0, ion strength = 0.05M). The average energy contribution is shown as dots, the standard deviation as bars. c, Per residue detail of Gα-GDP and Gα-GSP (non-hydrolyzable GTP analog) consensus contacts. The barplot shows the frequency of finding a contact mediated by topologically equivalent positions with GDP/GSP. Side-chain contacts are shown as dark grey bars, main-chain contacts as light grey bars. The degree of conservation of contacting residues (calculated from the 561 complete Gα sequences) is represented in the right panel and the consensus sequence for each position is shown.
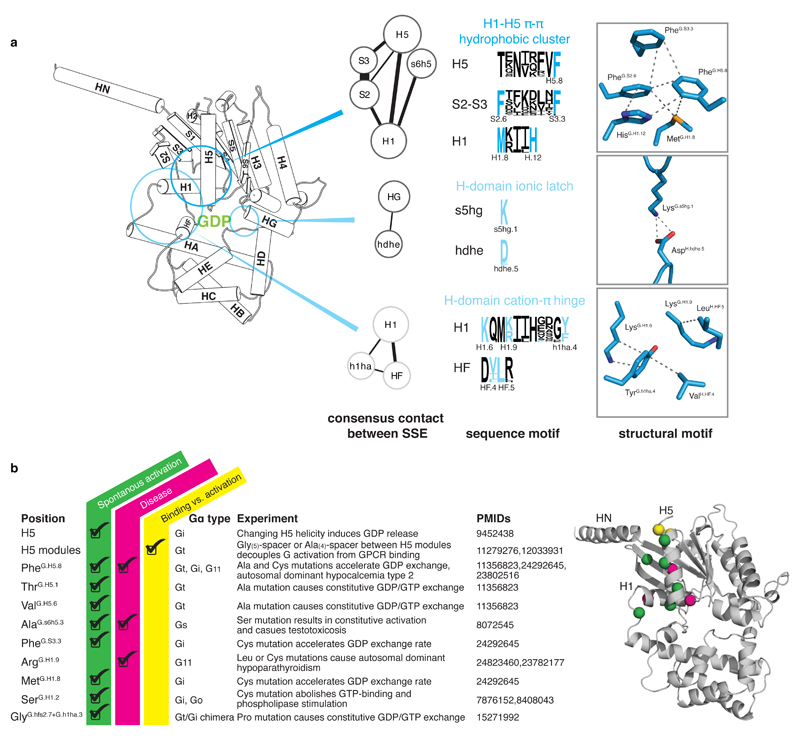
a, A universally conserved cluster of pi-pi and hydrophobic interactions between S2 (PheG.S2.6) and S3 (PheG.S3.3), H1 (MetG.H1.8 and HisG.H1.12) and H5 (PheG.H5.8) links H5 and H1 in the absence of the receptor. Upon receptor binding, residues within this motif (PheG.H5.8 and PheG.S3.3) interact with the conserved Pro and Leu of ICL2 of the receptor as has been shown for Gαs (3sn6) and Gαi (Mnpotra et al). Interrupting the contacts between H5 and H1 seems to be the trigger for transmitting the signal of GPCR binding to helix H1 (which interacts with GDP and the H-domain.) The only conserved residue contact between the H-domain and the G-domain that is not in the hinge region is formed by a universally conserved salt-bridge (H-domain ionic latch) between the very N-terminal end of HG of the G-domain (LysG.s5hg1) and the loop connecting HD and HE in the H domain (AspH.hdhe.5). The hinge region is formed by H1, the loop between H1 and HA, and HF. H1 interacts via (i) a cation-pi interaction mediated by a universally conserved residue with the loop connecting H1 and HA (LysG.H1.6 and TyrG.h1ha.4) and (ii) a hydrophobic interaction with HF (LysG.H1.9 and LeuH.HF.5). b, Disease and engineered mutations that can be explained by the universal Gα activation model mapped on a Gα protein. Cα position of residues are shown as spheres; mutations at green positions cause spontaneous GDP release by interrupting consensus contacts between conserved residues, thereby ‘mimicking’ the effect of receptor-binding to Gα. Pink positions have also been reported to cause disease by constitutively activating Gα. Insertion of an Ala4 or Gly5 after the yellow position separate the H5 transmission and interface module, thereby allowing GPCR-binding without triggering GDP release.
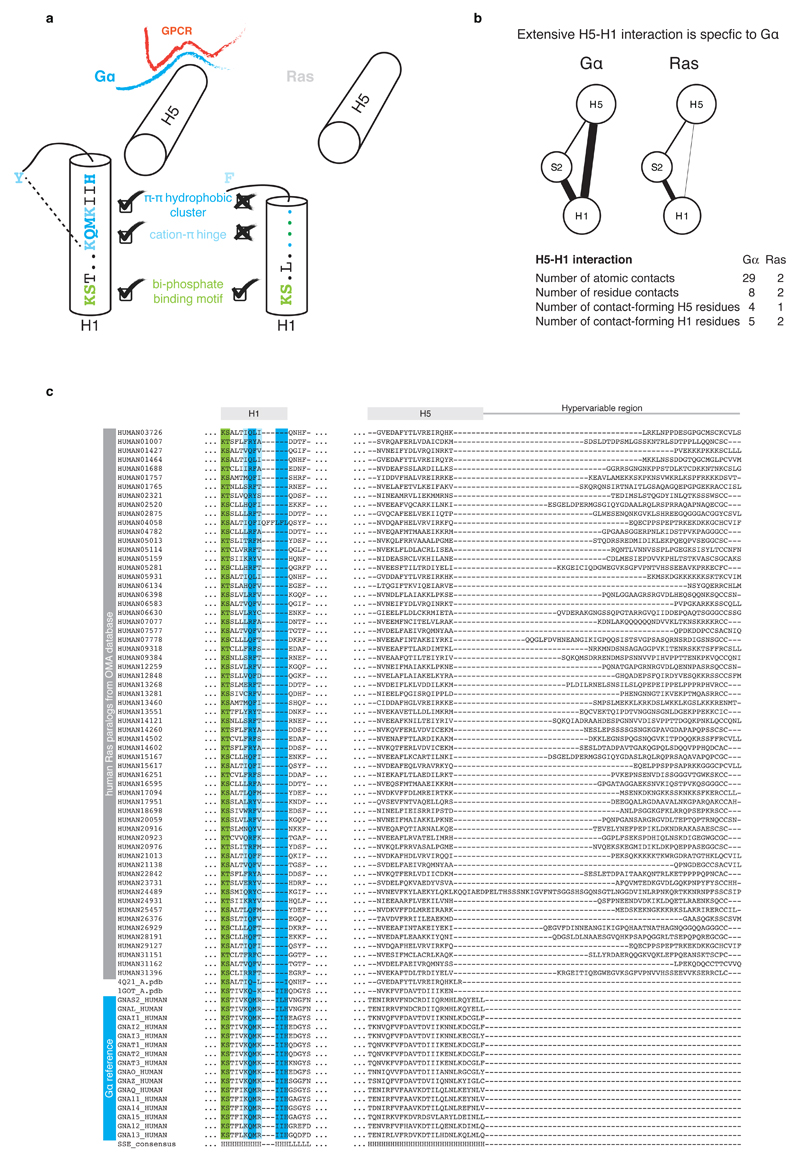
a, Schematic representation of structural motifs on H1 that are shared or unique to Gα and Ras. While the part of H1 with the phosphate-binding motif is conserved across both protein families, the C-terminal part is conserved only in Gα. H1 in Gα has three additional residues that allow for extensive residue contacts between H1 and H5. In Ras, these interactions are missing and H5 and H1 are both 3 residues shorter. The consensus sequence and secondary structure of equivalent residues of H1 in Gα and Ras is also depicted. b, Comparison of the residue contact network between topologically equivalent residues in H5 and H1 in the corresponding inactive GDP-bound states of Gα (PDB 1got) and Ras (PDB 4q21). The weight of the link between SSEs denotes the number of atomic contacts. c, Sequence alignments of H1 and H5 of human Gα and Ras paralogs. The sequence alignment was obtained based on cross-referencing the alignments using the structures of Gα and Ras.
