

Salt Sensitivity in Response to Renal Injury Requires Renal Angiotensin-Converting Enzyme
- PMID: 26150439
- PMCID: PMC4825680
- DOI: 10.1161/HYPERTENSIONAHA.115.05320
Salt Sensitivity in Response to Renal Injury Requires Renal Angiotensin-Converting Enzyme
Abstract
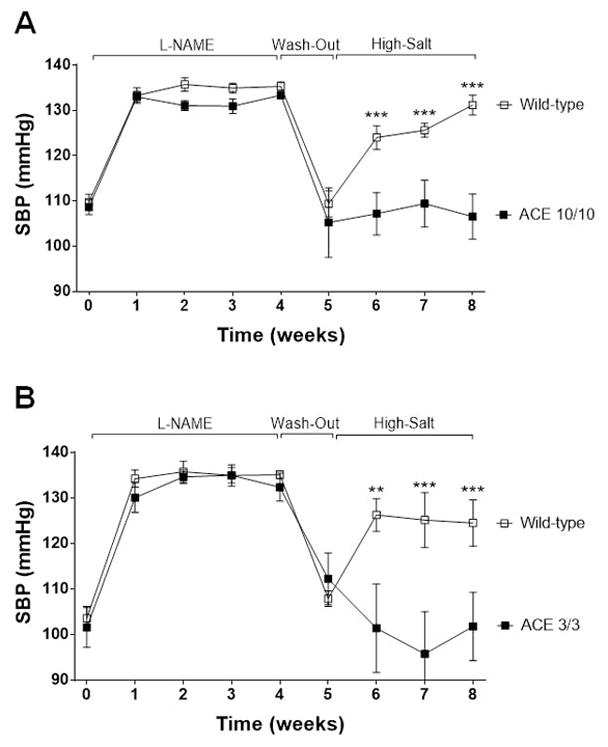
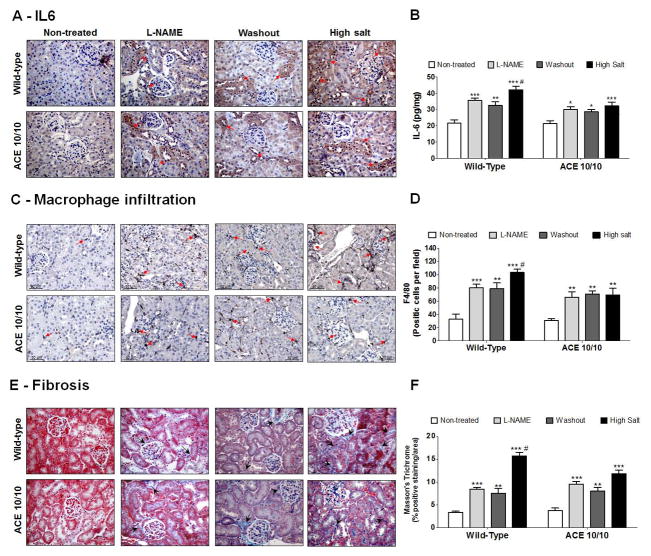
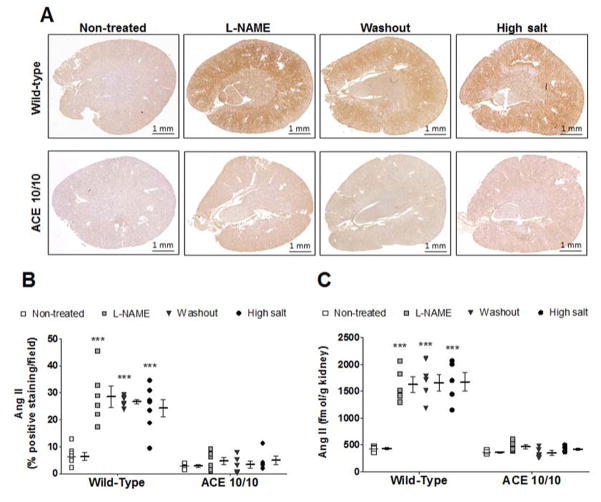
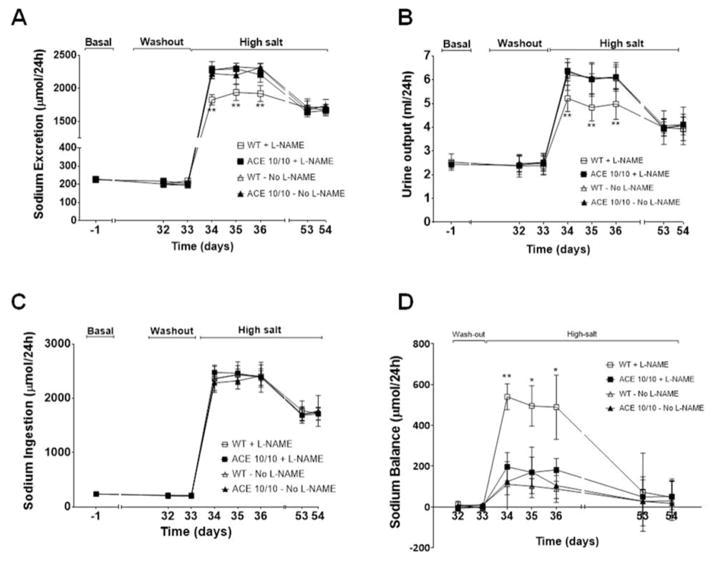
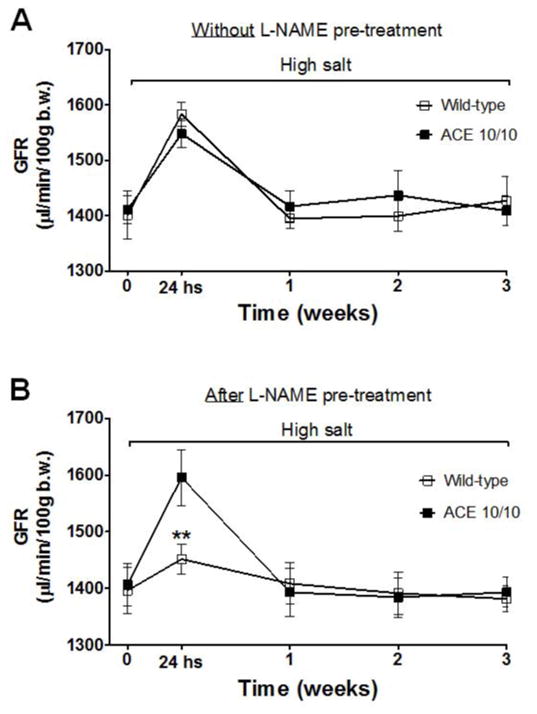
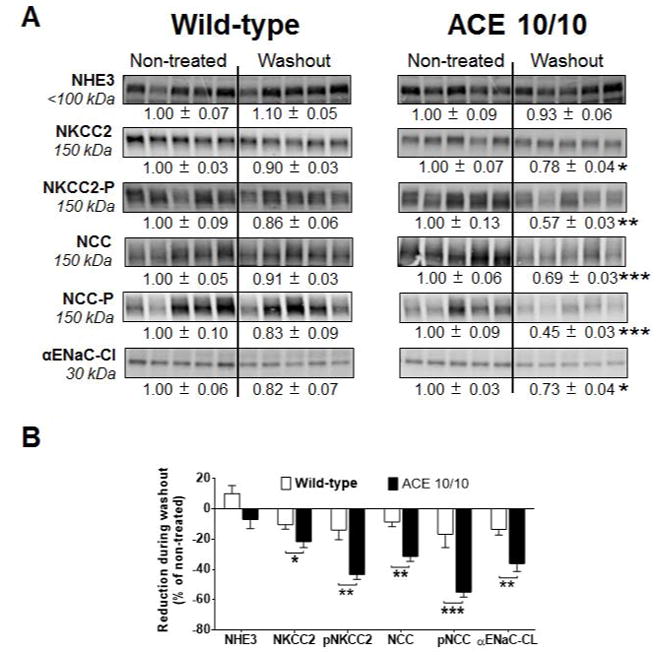
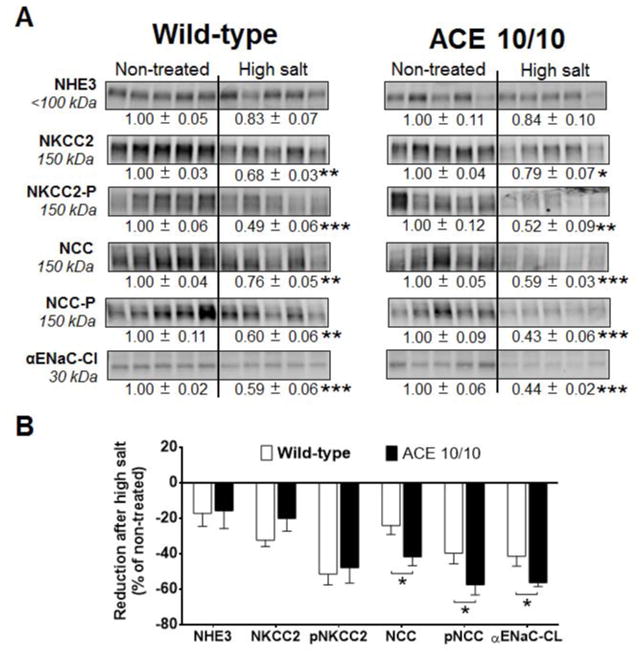
Recent evidence indicates that salt-sensitive hypertension can result from a subclinical injury that impairs the kidneys' capacity to properly respond to a high-salt diet. However, how this occurs is not well understood. Here, we showed that although previously salt-resistant wild-type mice became salt sensitive after the induction of renal injury with the nitric oxide synthase inhibitor Nω-nitro-l-arginine methyl ester hydrochloride; mice lacking renal angiotensin-converting enzyme, exposed to the same insult, did not become hypertensive when faced with a sodium load. This is because the activity of renal angiotensin-converting enzyme plays a critical role in (1) augmenting the local pool of angiotensin II and (2) the establishment of the antinatriuretic state via modulation of glomerular filtration rate and sodium tubular transport. Thus, this study demonstrates that the presence of renal angiotensin-converting enzyme plays a pivotal role in the development of salt sensitivity in response to renal injury.
Keywords: angiotensin-converting enzyme; diet; glomerular filtration rate; hypertension; inflammation.
© 2015 American Heart Association, Inc.
Figures
References
-
- Lifton RP. Molecular genetics of human blood pressure variation. Science. 1996;272:676–680. - PubMed
-
- Franco M, Tapia E, Bautista R, Pacheco U, Santamaria J, Quiroz Y, Johnson RJ, Rodriguez-Iturbe B. Impaired pressure natriuresis resulting in salt-sensitive hypertension is caused by tubulointerstitial immune cell infiltration in the kidney. American journal of physiology. Renal physiology. 2013;304:F982–990. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
