

EGFR inhibition evokes innate drug resistance in lung cancer cells by preventing Akt activity and thus inactivating Ets-1 function
- PMID: 26150526
- PMCID: PMC4517222
- DOI: 10.1073/pnas.1510733112
EGFR inhibition evokes innate drug resistance in lung cancer cells by preventing Akt activity and thus inactivating Ets-1 function
Abstract
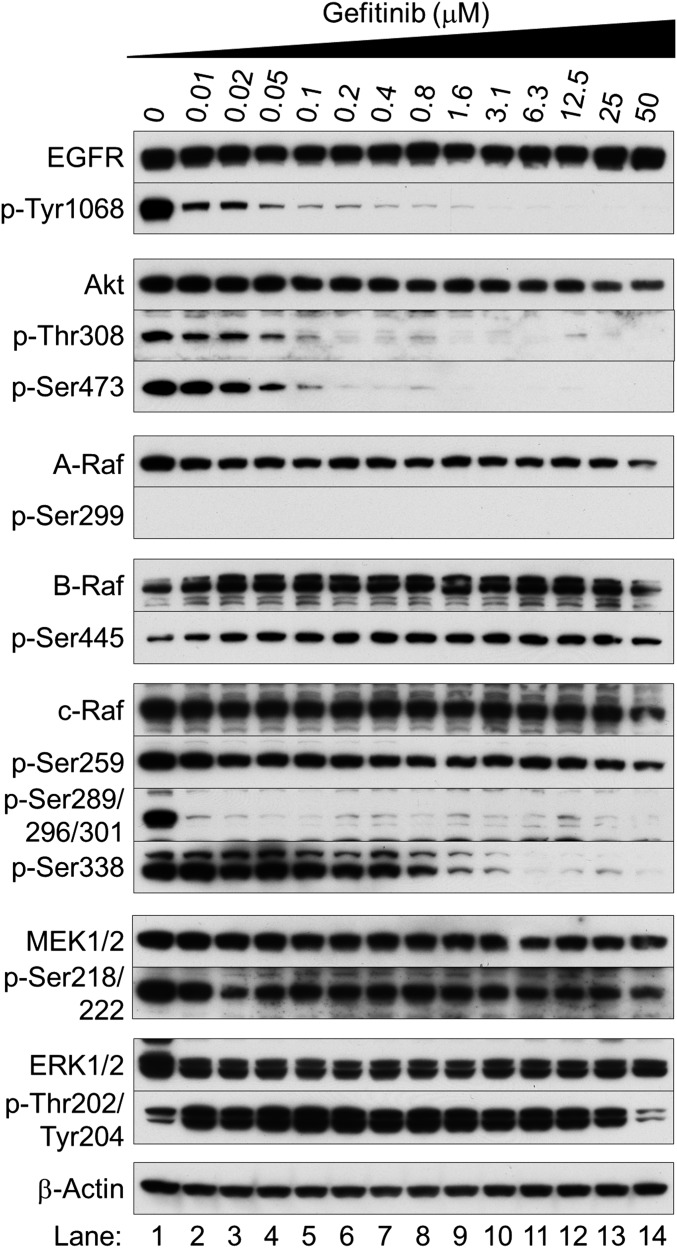
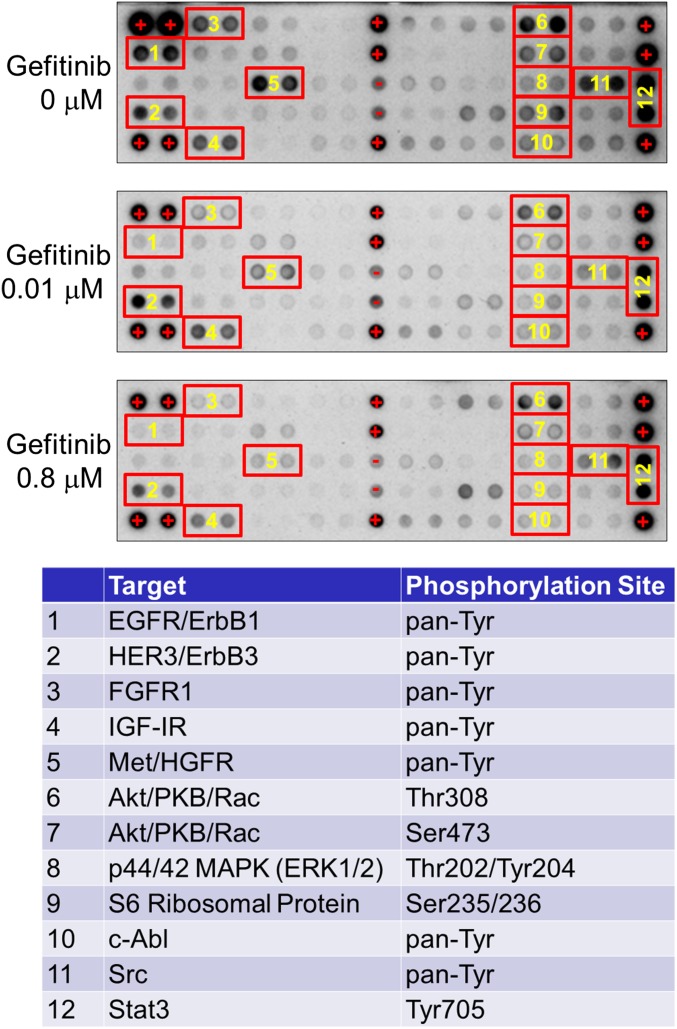
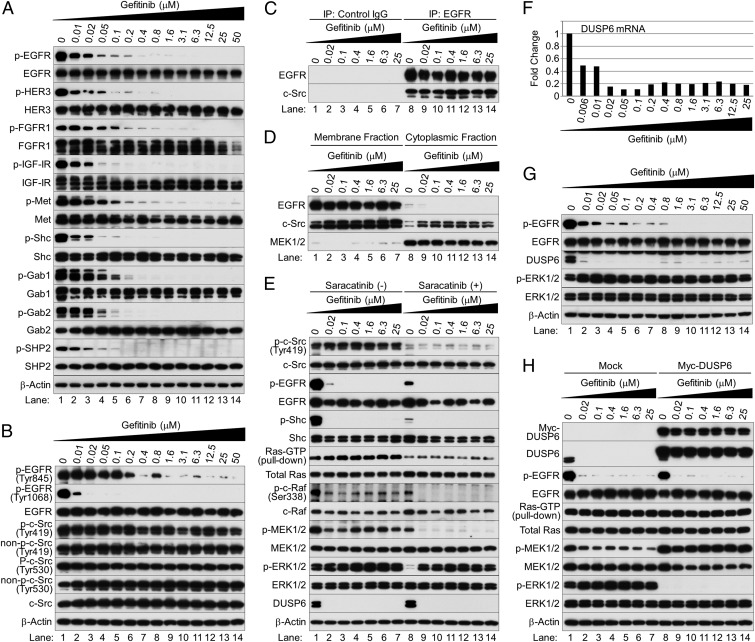
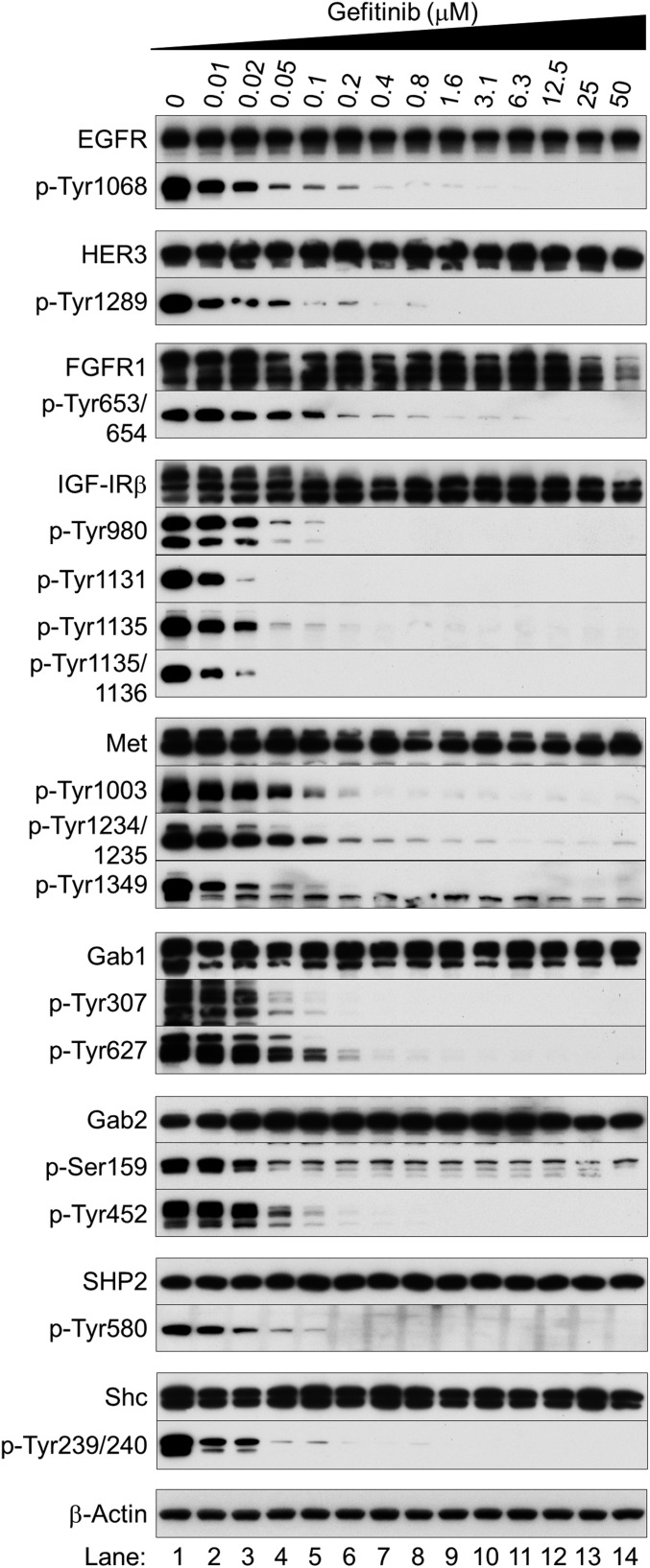
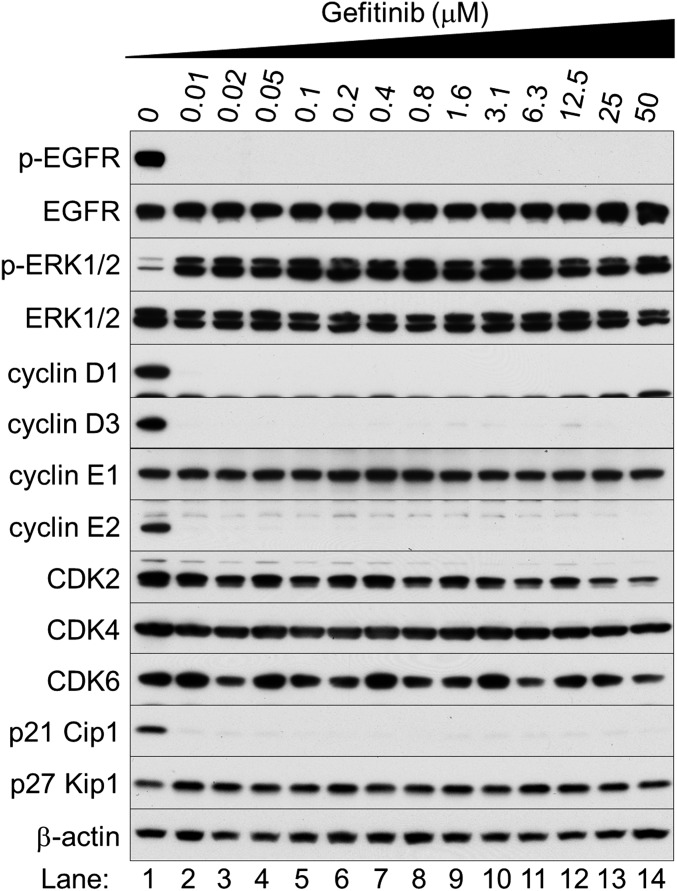
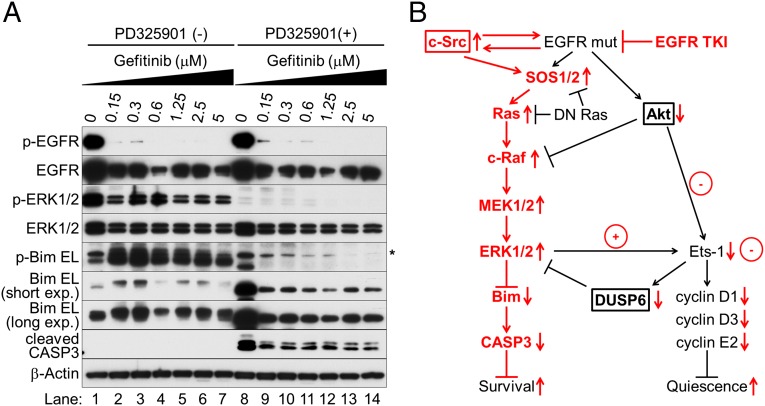
Nonsmall cell lung cancer (NSCLC) is the leading cause of cancer death worldwide. About 14% of NSCLCs harbor mutations in epidermal growth factor receptor (EGFR). Despite remarkable progress in treatment with tyrosine kinase inhibitors (TKIs), only 5% of patients achieve tumor reduction >90%. The limited primary responses are attributed partly to drug resistance inherent in the tumor cells before therapy begins. Recent reports showed that activation of receptor tyrosine kinases (RTKs) is an important determinant of this innate drug resistance. In contrast, we demonstrate that EGFR inhibition promotes innate drug resistance despite blockade of RTK activity in NSCLC cells. EGFR TKIs decrease both the mitogen-activated protein kinase (MAPK) and Akt protein kinase pathways for a short time, after which the Ras/MAPK pathway becomes reactivated. Akt inhibition selectively blocks the transcriptional activation of Ets-1, which inhibits its target gene, dual specificity phosphatase 6 (DUSP6), a negative regulator specific for ERK1/2. As a result, ERK1/2 is activated. Furthermore, elevated c-Src stimulates Ras GTP-loading and activates Raf and MEK kinases. These observations suggest that not only ERK1/2 but also Akt activity is essential to maintain Ets-1 in an active state. Therefore, despite high levels of ERK1/2, Ets-1 target genes including DUSP6 and cyclins D1, D3, and E2 remain suppressed by Akt inhibition. Reduction of DUSP6 in combination with elevated c-Src renews activation of the Ras/MAPK pathway, which enhances cell survival by accelerating Bim protein turnover. Thus, EGFR TKIs evoke innate drug resistance by preventing Akt activity and inactivating Ets-1 function in NSCLC cells.
Keywords: EGFR inhibition; ERK1/2 paradoxical activation; innate drug resistance; nonsmall cell lung cancer; tyrosine kinase inhibitors.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
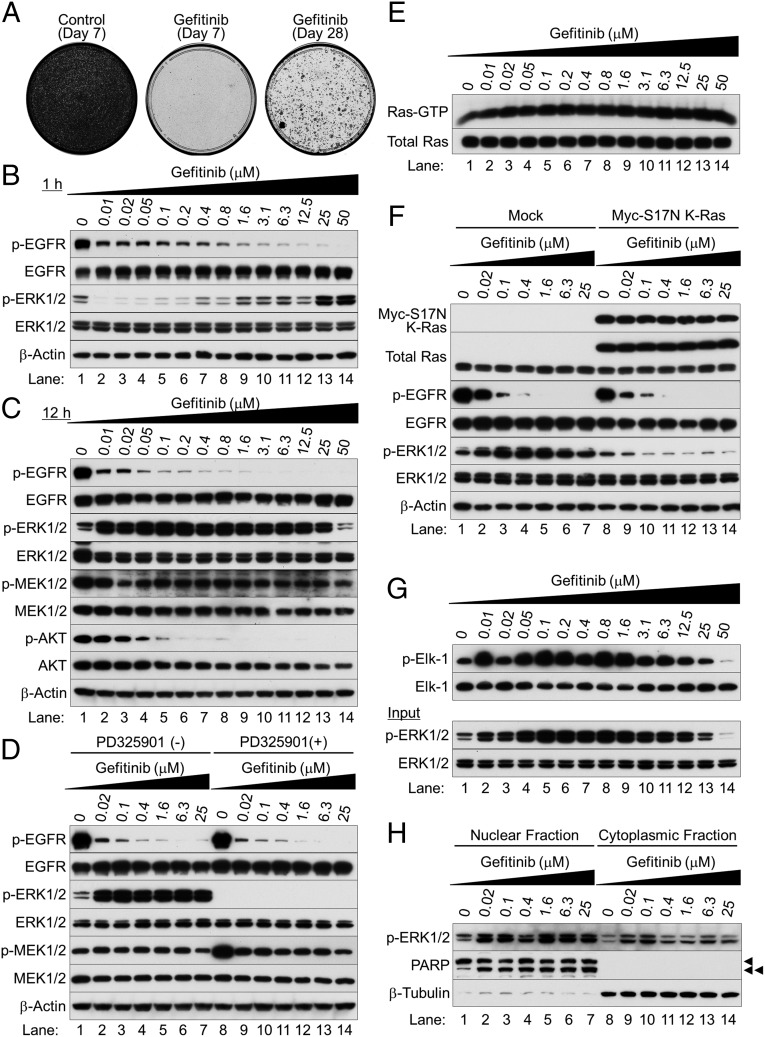
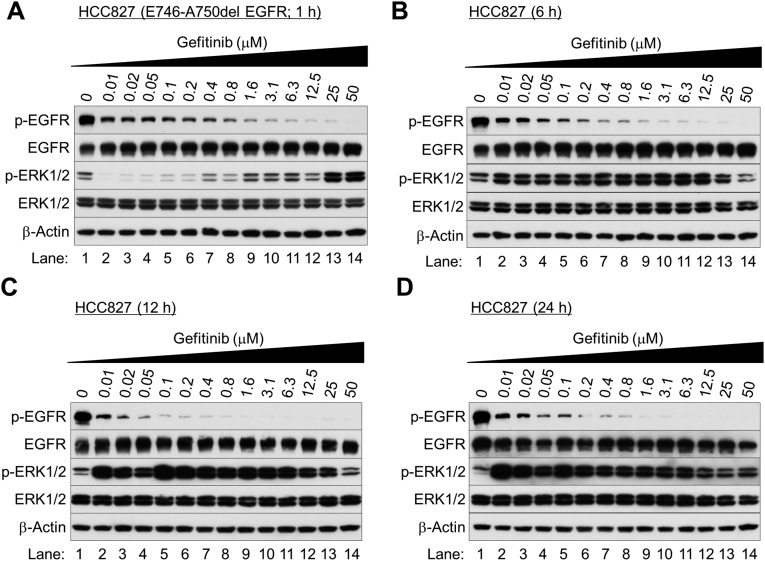
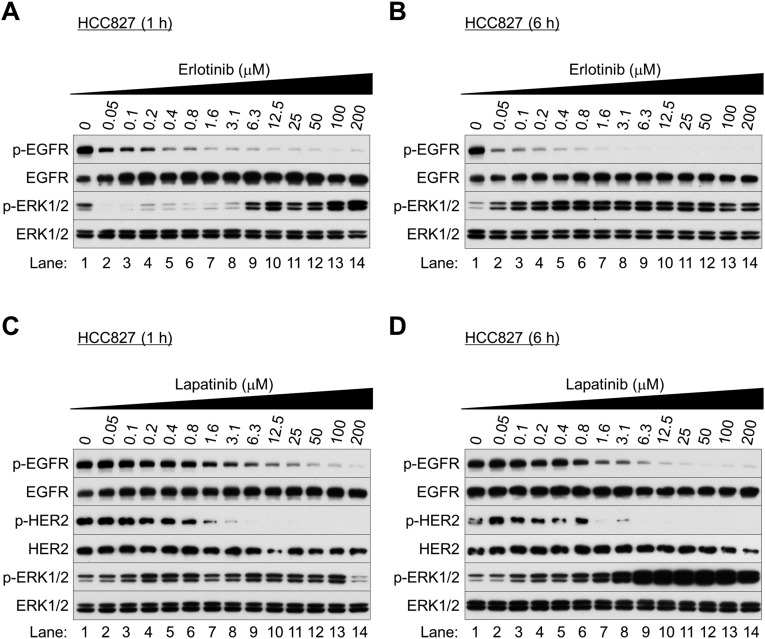
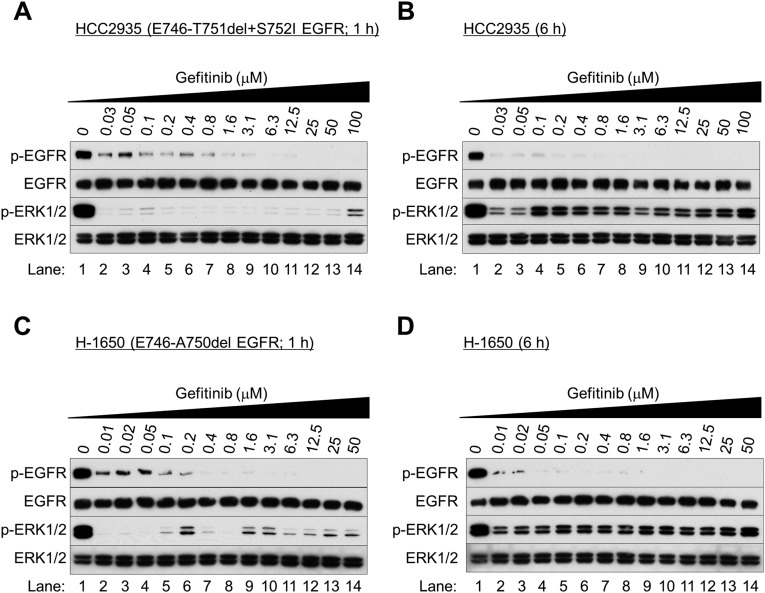


Comment in
-
Resistance to EGFR-targeted therapy by Ets-1 inactivation.Cell Cycle. 2015;14(20):3211-2. doi: 10.1080/15384101.2015.1086200. Cell Cycle. 2015. PMID: 26313421 Free PMC article. No abstract available.
References
-
- Rosell R, et al. Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
