

Progesterone receptor modulates ERα action in breast cancer
- PMID: 26153859
- PMCID: PMC4650274
- DOI: 10.1038/nature14583
Progesterone receptor modulates ERα action in breast cancer
Erratum in
-
Corrigendum: Progesterone receptor modulates ERα action in breast cancer.Nature. 2015 Oct 1;526(7571):144. doi: 10.1038/nature14959. Epub 2015 Aug 5. Nature. 2015. PMID: 26245370 No abstract available.
Abstract
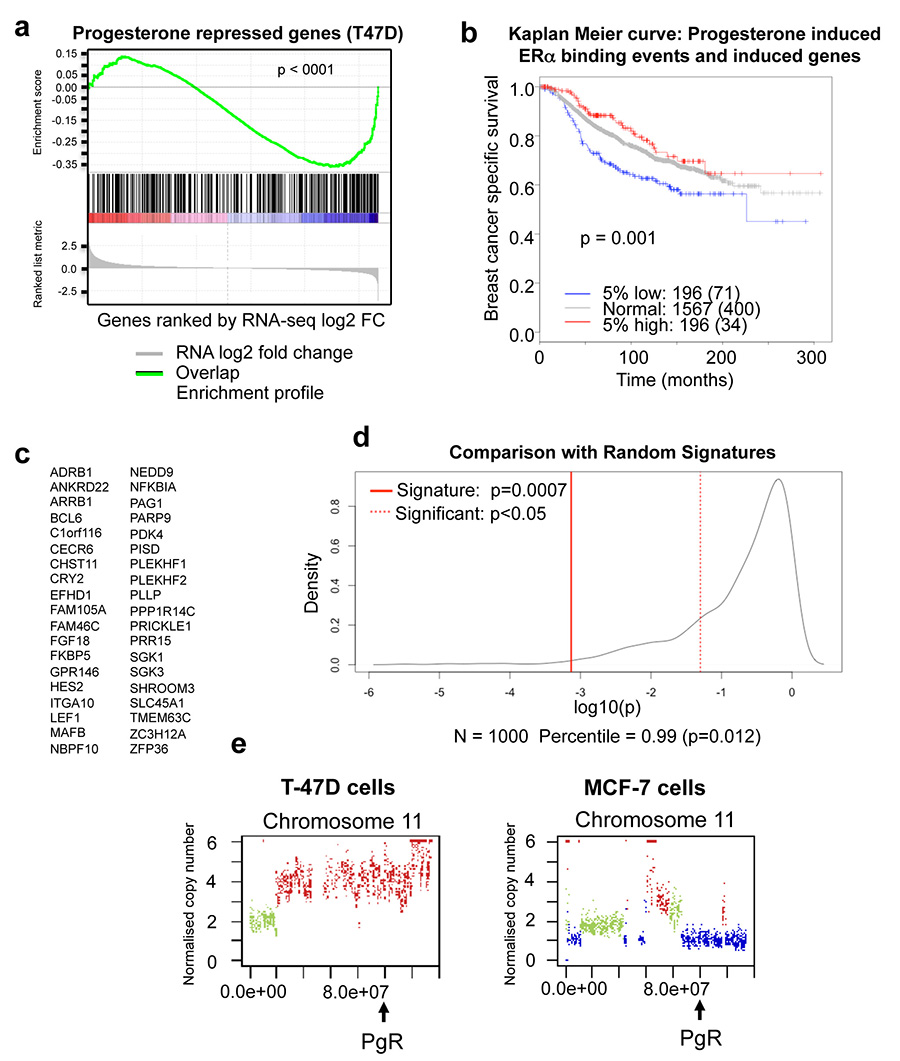
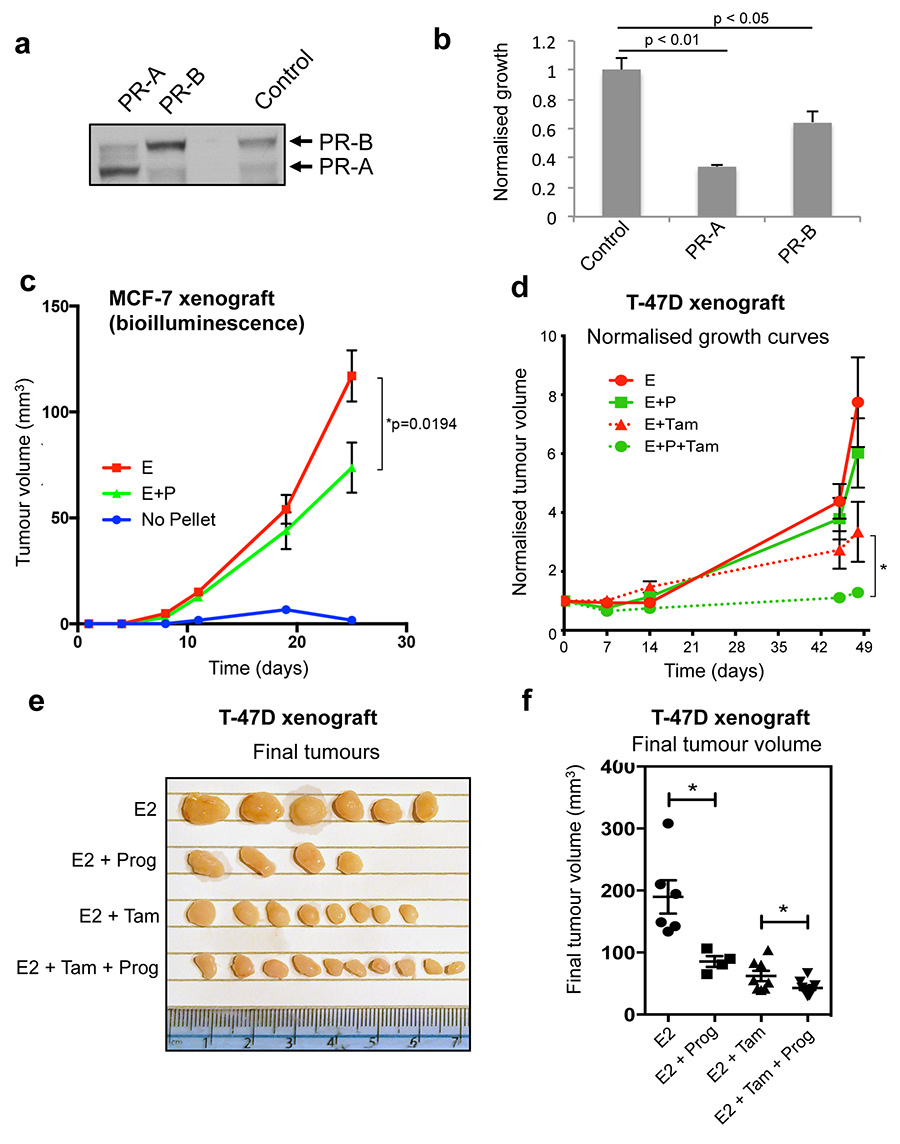
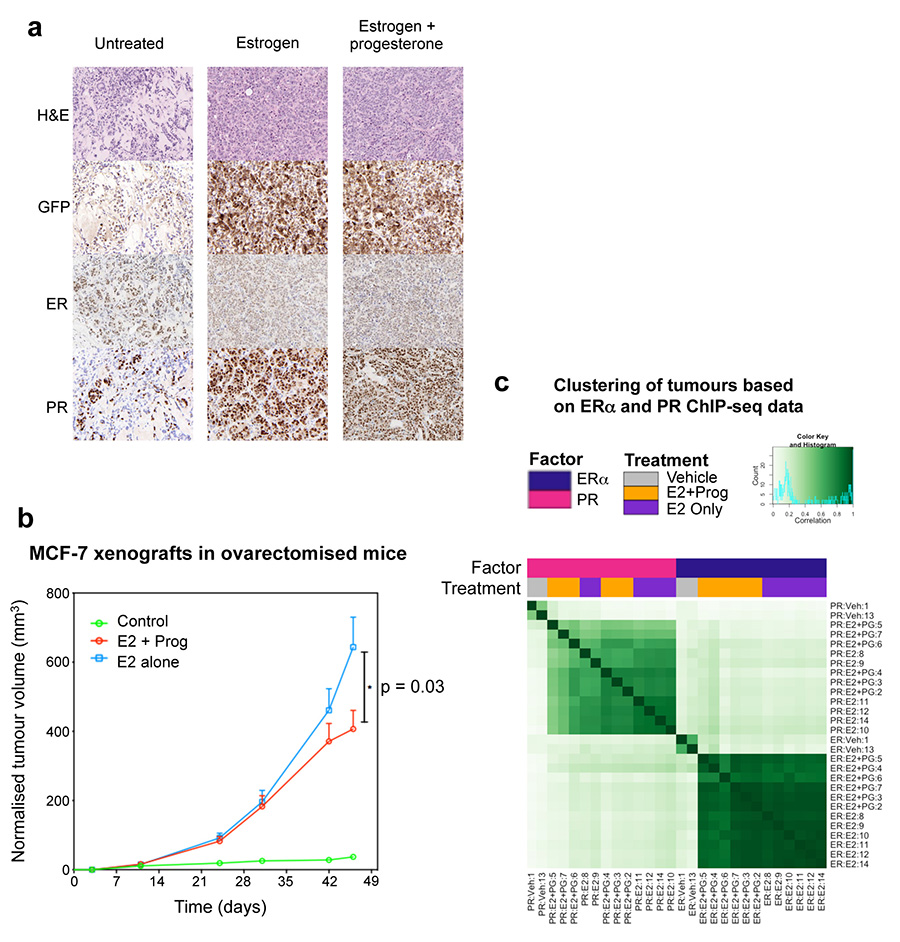
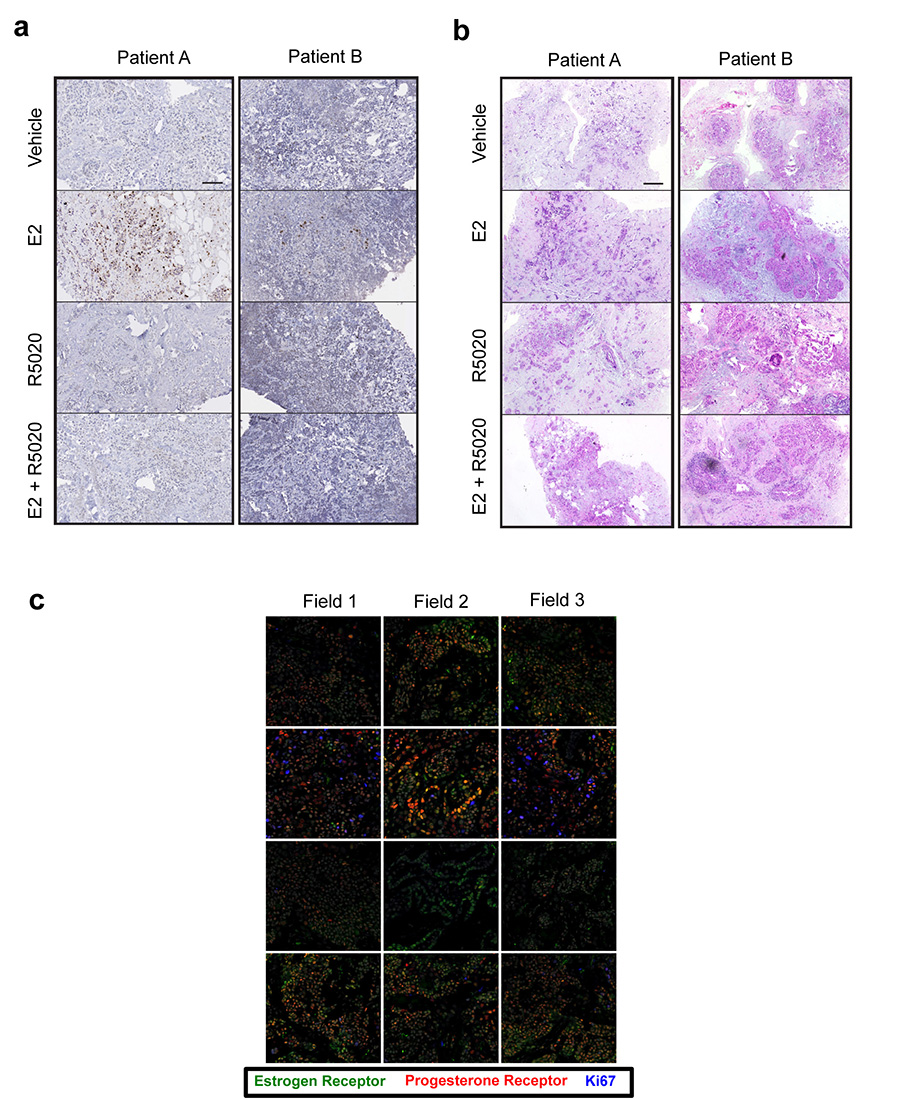
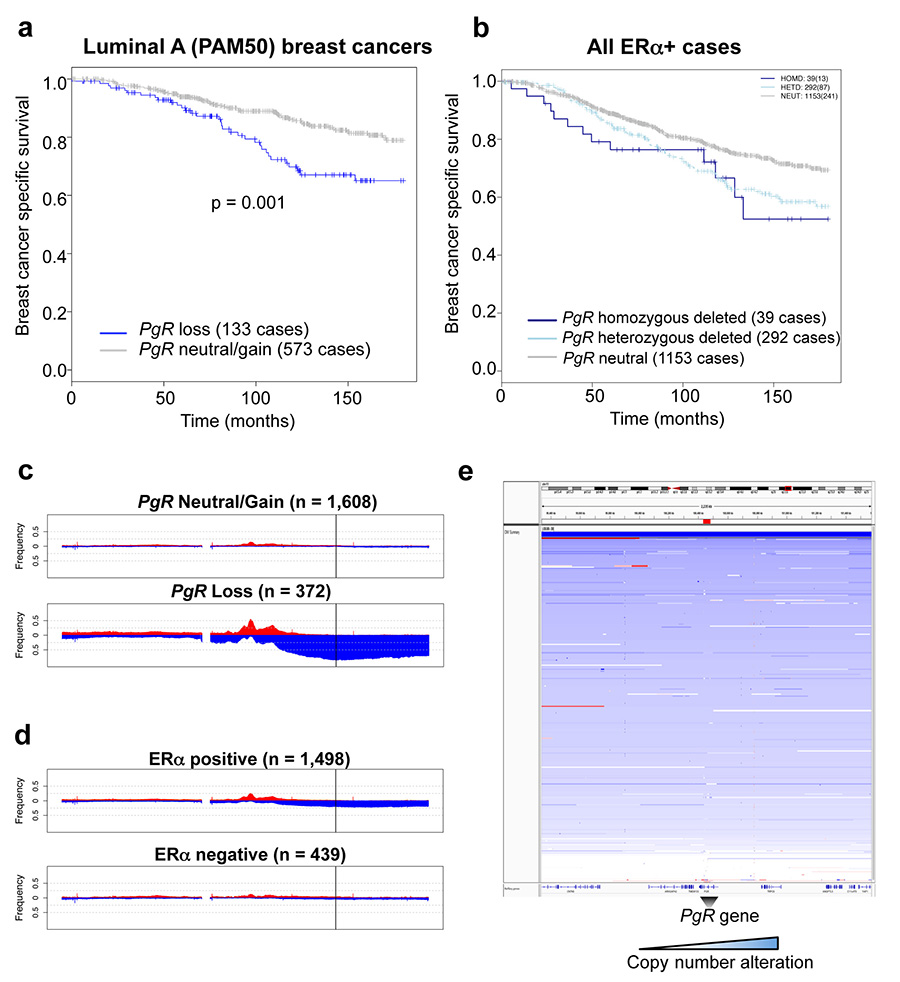
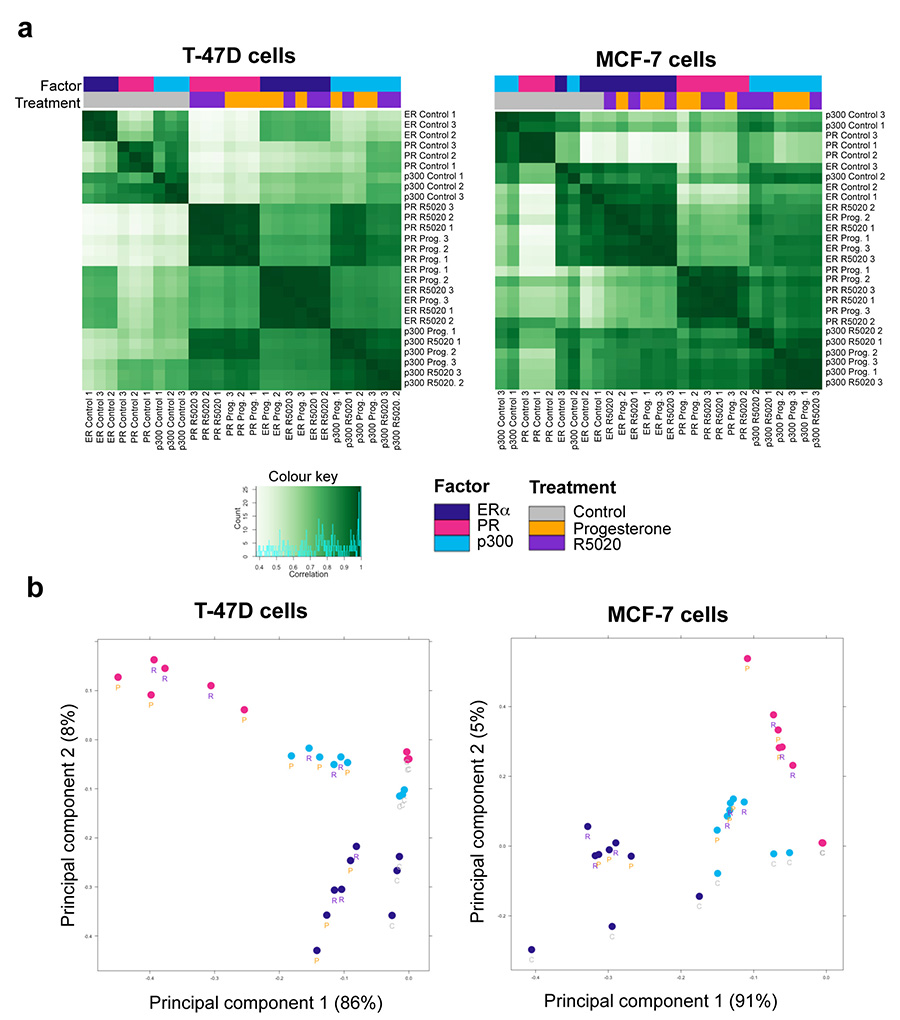
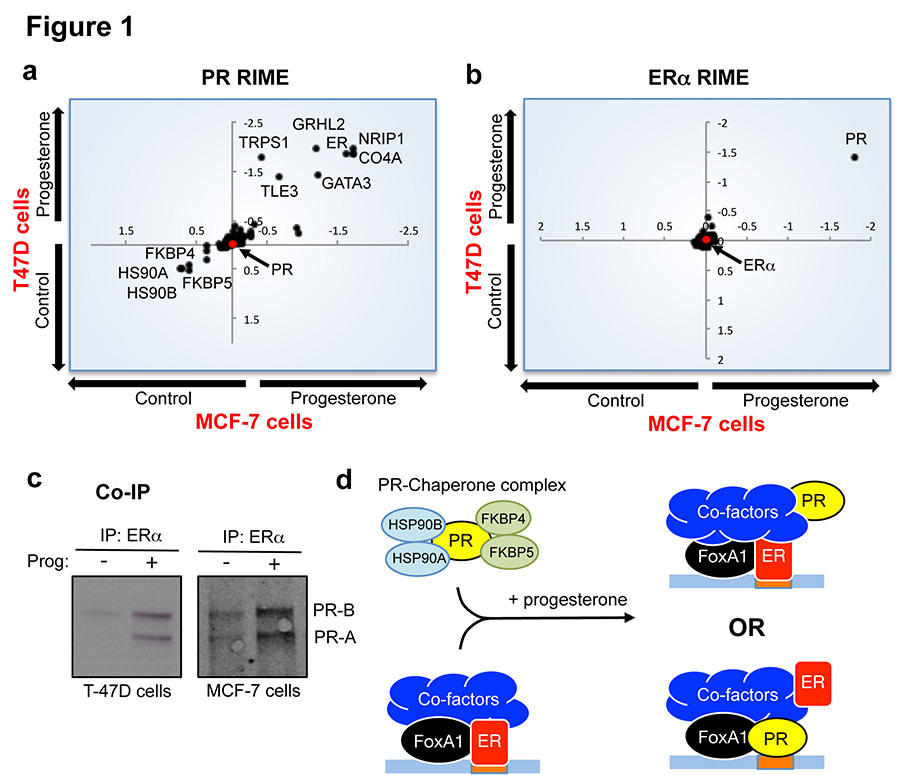
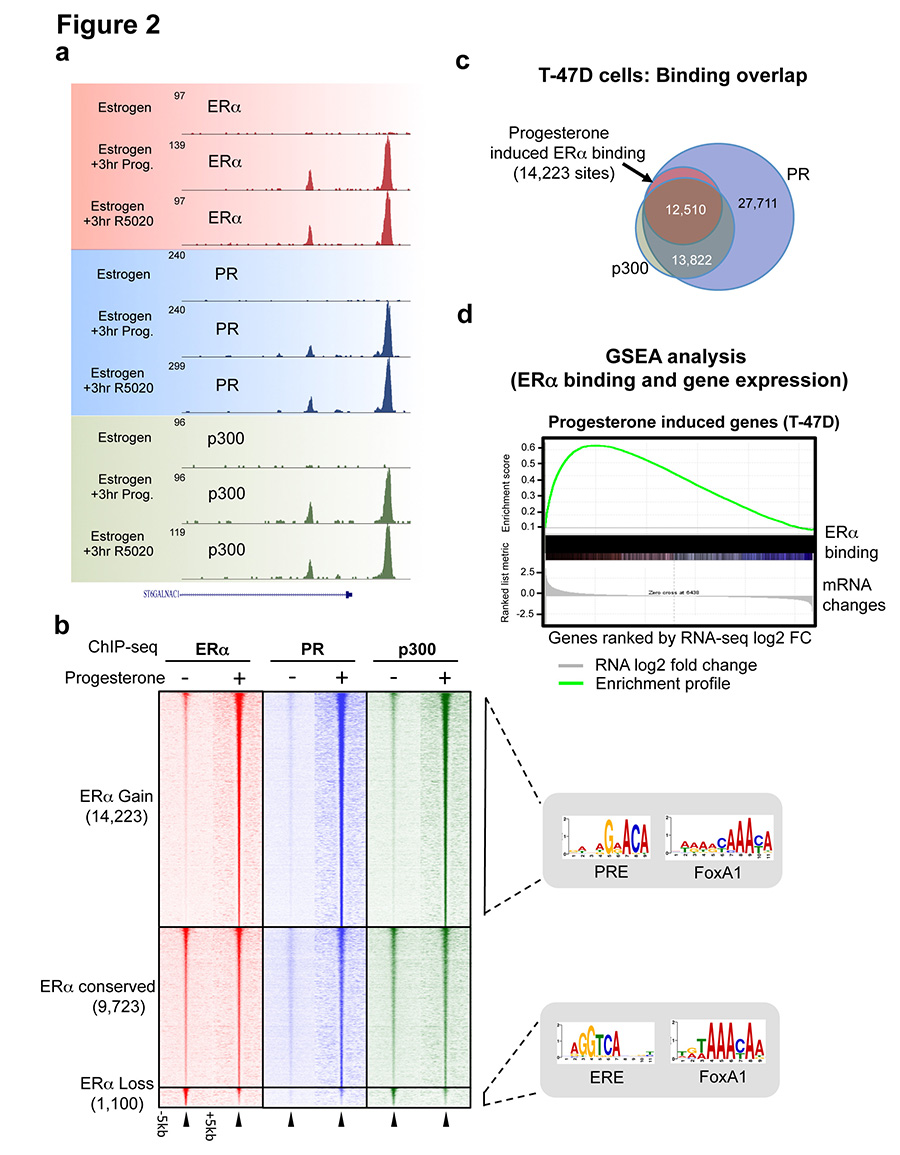
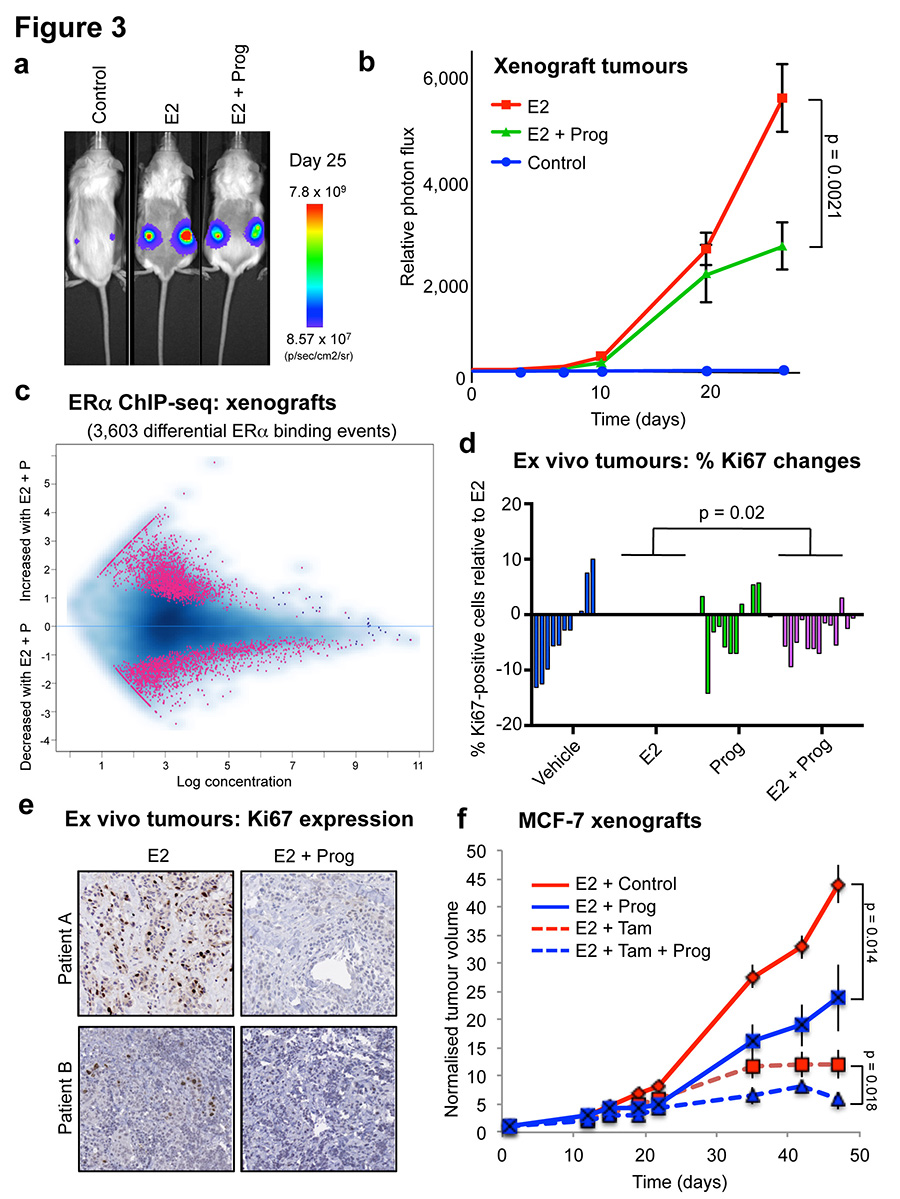
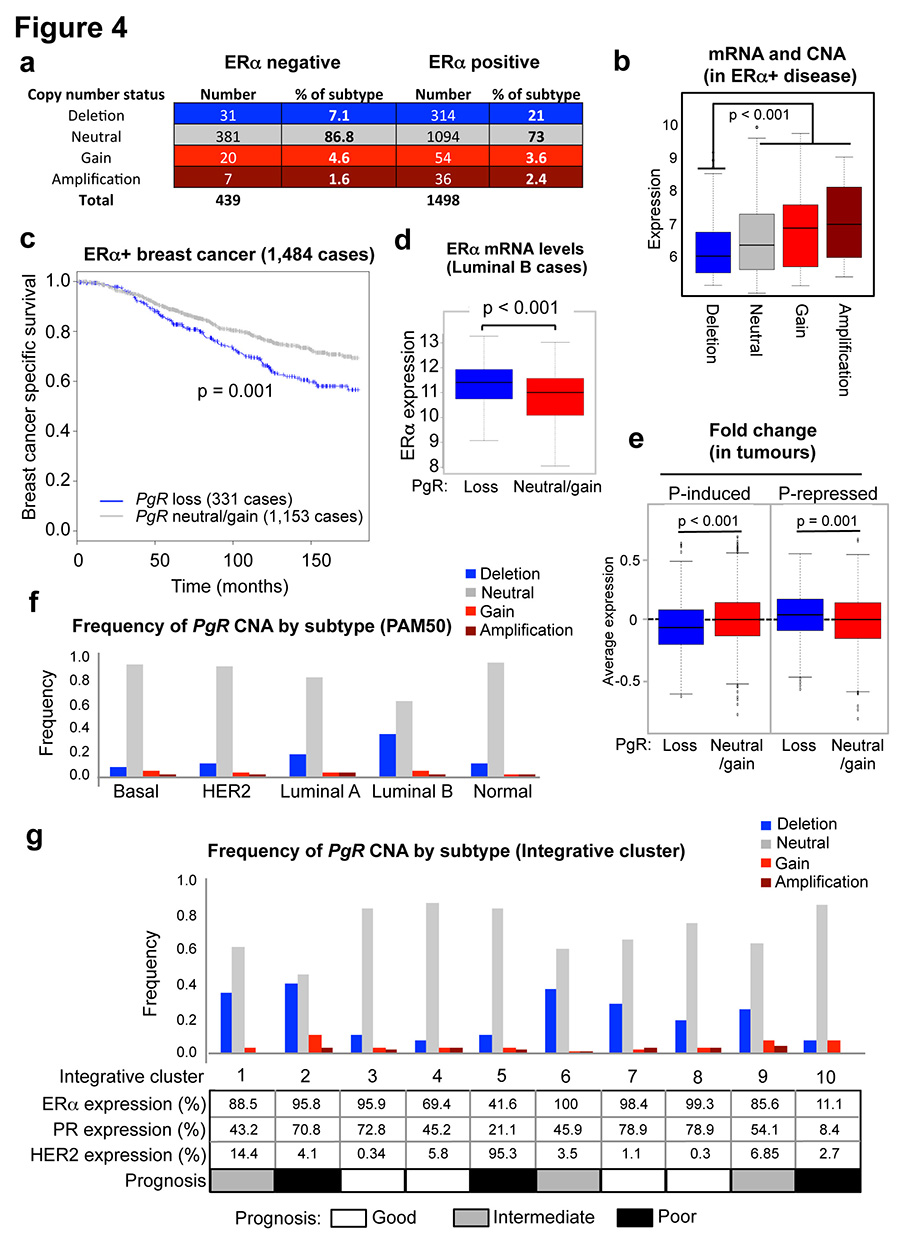
Progesterone receptor (PR) expression is used as a biomarker of oestrogen receptor-α (ERα) function and breast cancer prognosis. Here we show that PR is not merely an ERα-induced gene target, but is also an ERα-associated protein that modulates its behaviour. In the presence of agonist ligands, PR associates with ERα to direct ERα chromatin binding events within breast cancer cells, resulting in a unique gene expression programme that is associated with good clinical outcome. Progesterone inhibited oestrogen-mediated growth of ERα(+) cell line xenografts and primary ERα(+) breast tumour explants, and had increased anti-proliferative effects when coupled with an ERα antagonist. Copy number loss of PGR, the gene coding for PR, is a common feature in ERα(+) breast cancers, explaining lower PR levels in a subset of cases. Our findings indicate that PR functions as a molecular rheostat to control ERα chromatin binding and transcriptional activity, which has important implications for prognosis and therapeutic interventions.
Figures
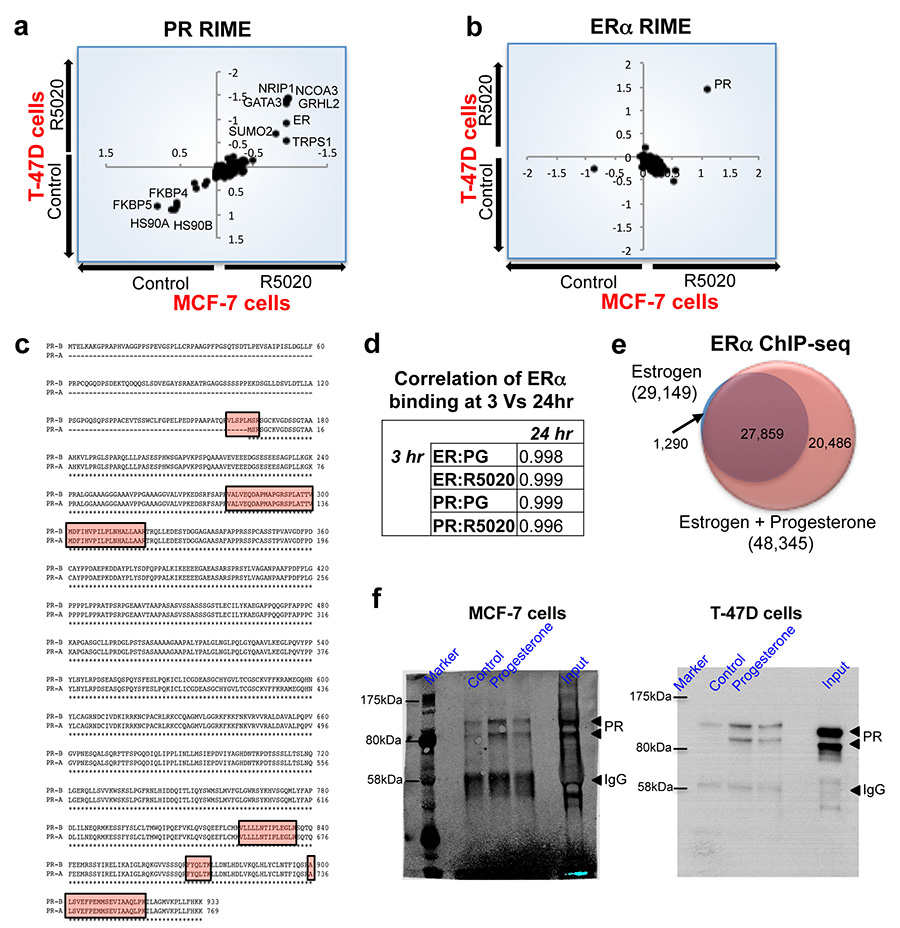
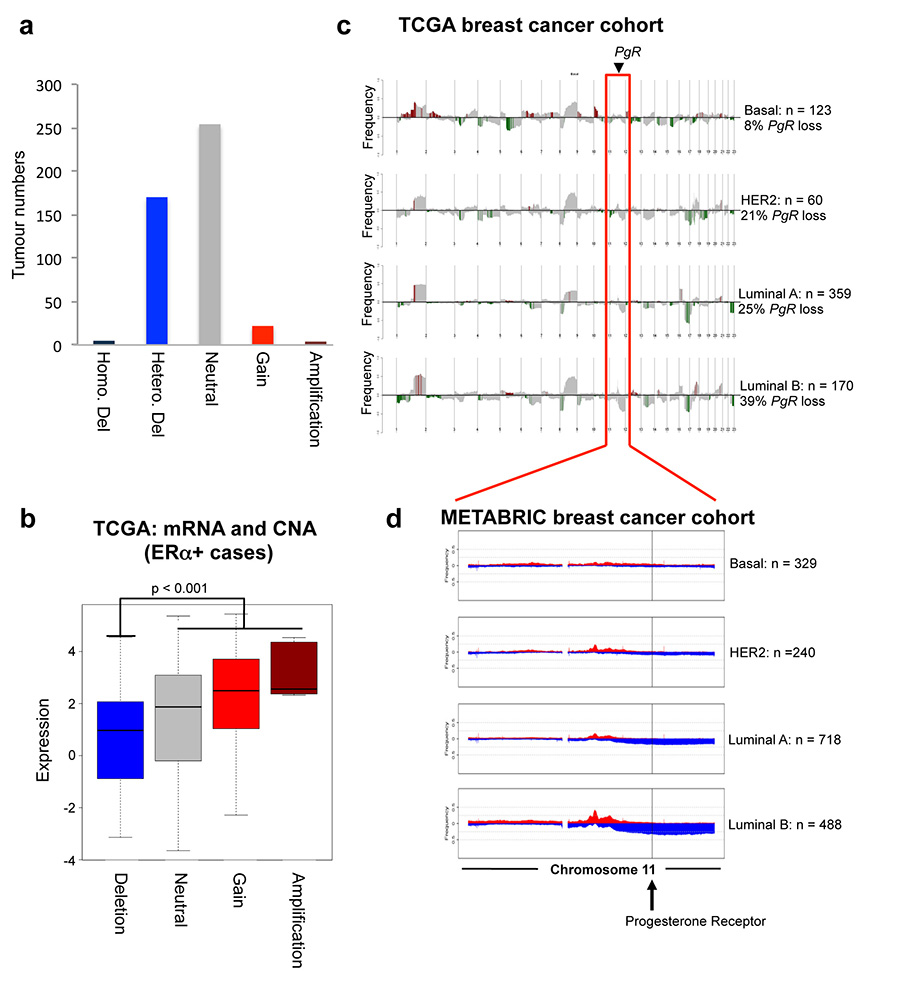
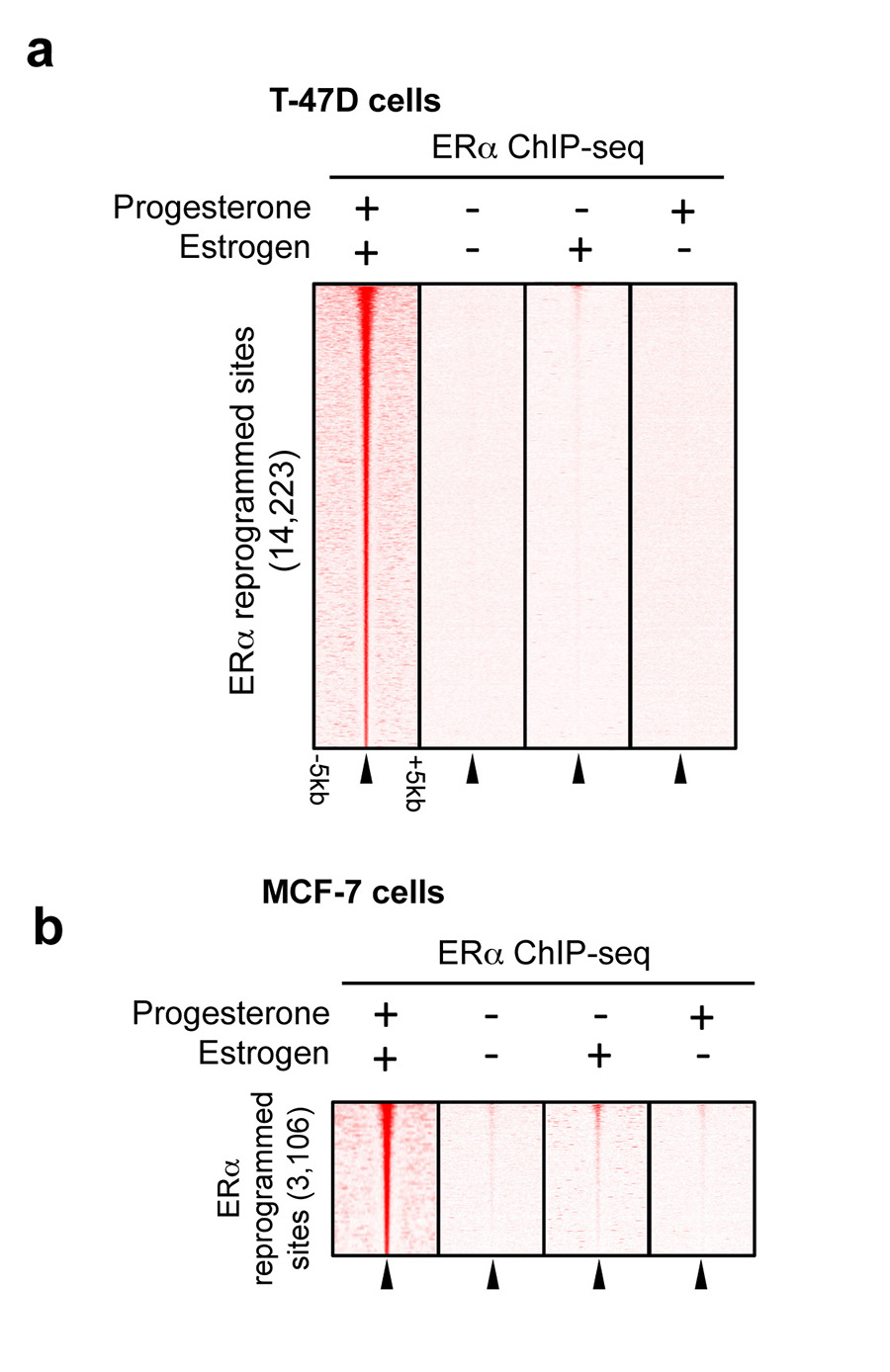
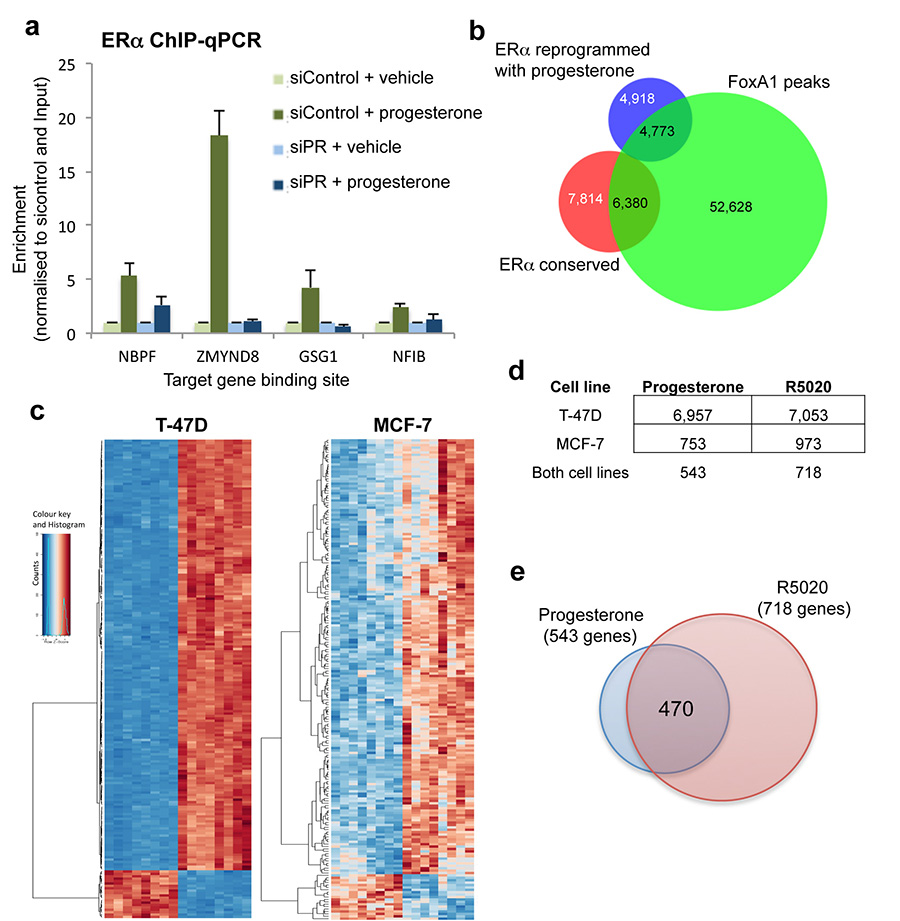
Comment in
-
Breast cancer: Untangling the role of progesterone receptors.Nat Rev Cancer. 2015 Aug;15(8):456. doi: 10.1038/nrc3991. Nat Rev Cancer. 2015. PMID: 26205336 No abstract available.
References
-
- Bentel JM, et al. Androgen receptor agonist activity of the synthetic progestin, medroxyprogesterone acetate, in human breast cancer cells. Mol Cell Endocrinol. 1999;154:11–20. doi:S0303-7207(99)00109-4 [pii] - PubMed
-
- Vignon F, Bardon S, Chalbos D, Rochefort H. Antiestrogenic effect of R5020, a synthetic progestin in human breast cancer cells in culture. J Clin Endocrinol Metab. 1983;56:1124–1130. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
