

Strategies for fine-mapping complex traits
- PMID: 26157023
- PMCID: PMC4572002
- DOI: 10.1093/hmg/ddv260
Strategies for fine-mapping complex traits
Abstract
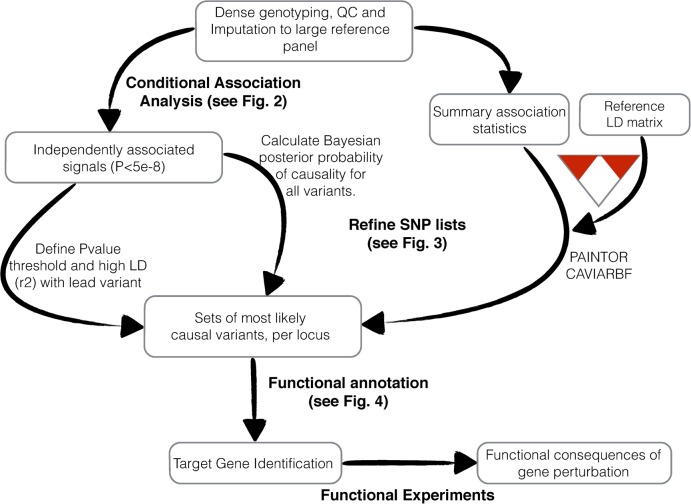
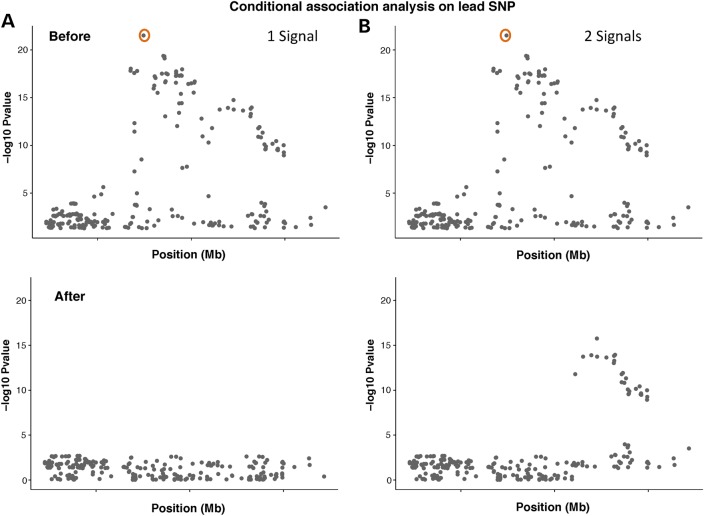
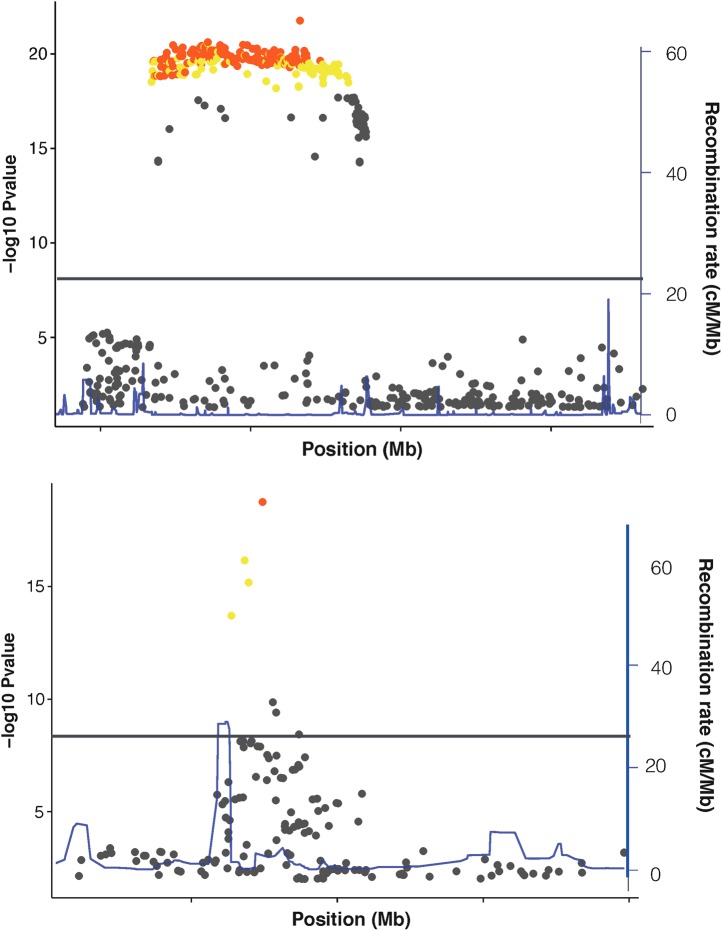
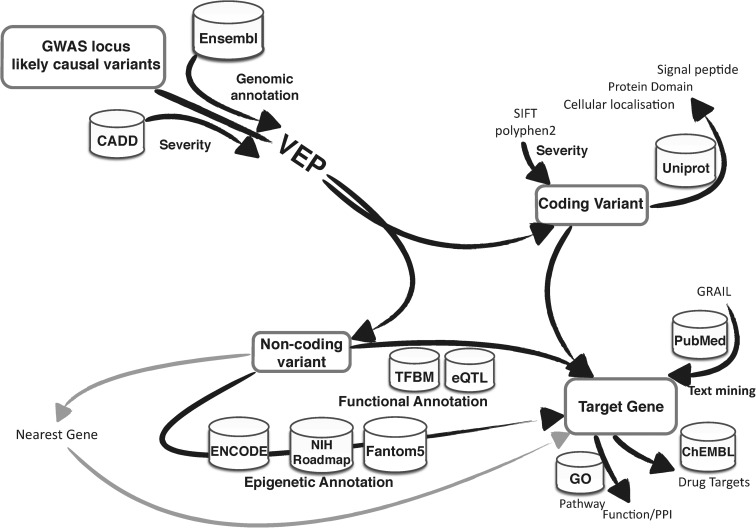
Genome-wide association studies (GWAS) have identified thousands of robust and replicable genetic associations for complex disease. However, the identification of the causal variants that underlie these associations has been more difficult. This problem of fine-mapping association signals predates GWAS, but the last few years have seen a surge of studies aimed at pinpointing causal variants using both statistical evidence from large association data sets and functional annotations of genetic variants. Combining these two approaches can often determine not only the causal variant but also the target gene. Recent contributions include analyses of custom genotyping arrays, such as the Immunochip, statistical methods to identify credible sets of causal variants and the addition of functional genomic annotations for coding and non-coding variation to help prioritize variants and discern functional consequence and hence the biological basis of disease risk.
© The Author 2015. Published by Oxford University Press.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
