

Heterogeneous Network Edge Prediction: A Data Integration Approach to Prioritize Disease-Associated Genes
- PMID: 26158728
- PMCID: PMC4497619
- DOI: 10.1371/journal.pcbi.1004259
Heterogeneous Network Edge Prediction: A Data Integration Approach to Prioritize Disease-Associated Genes
Abstract
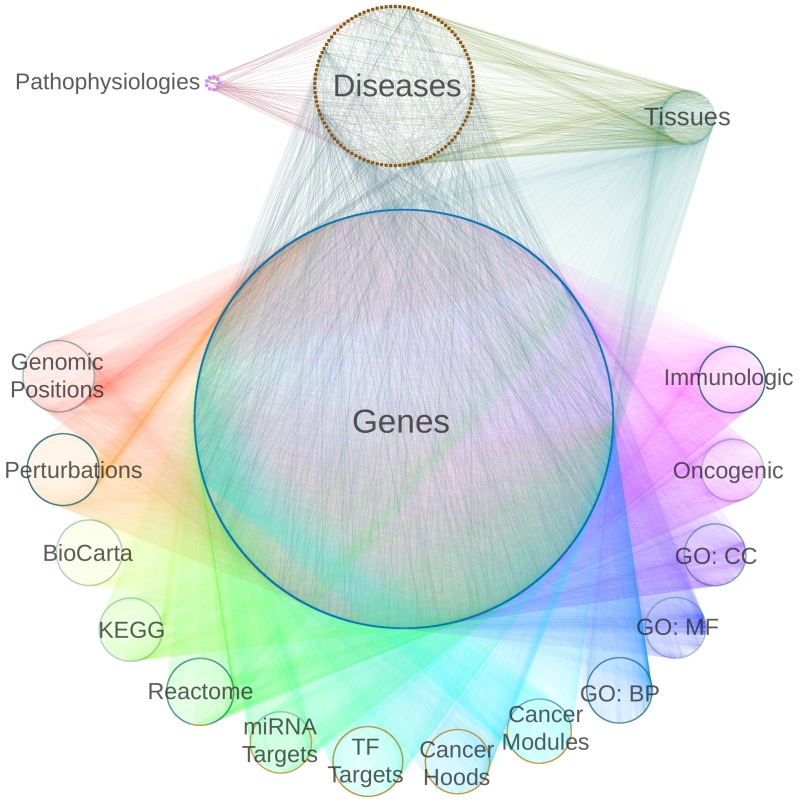
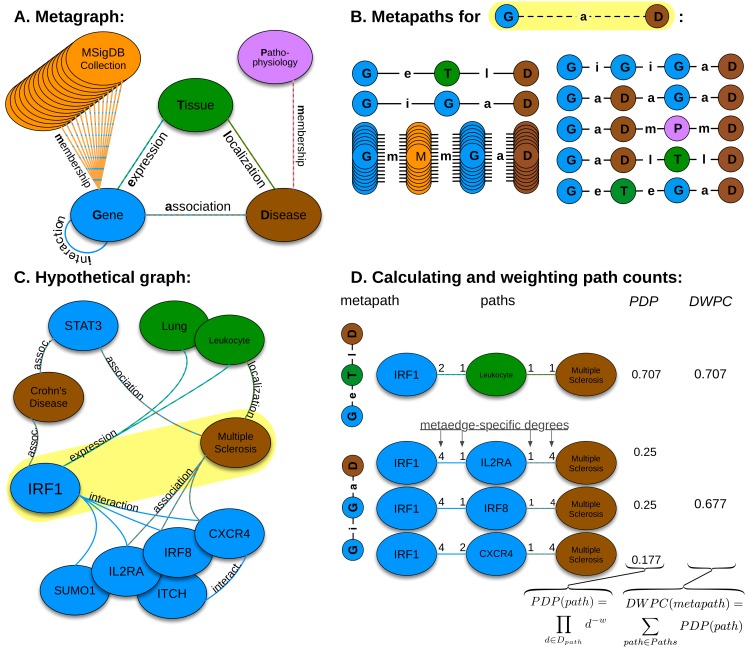
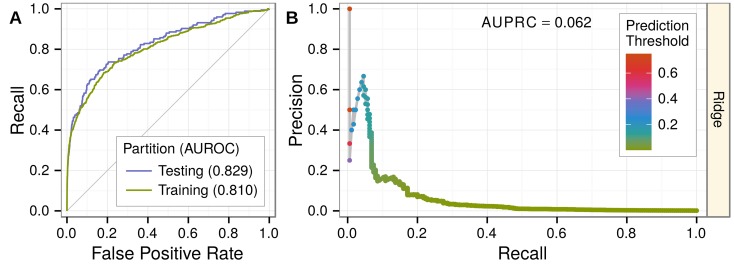
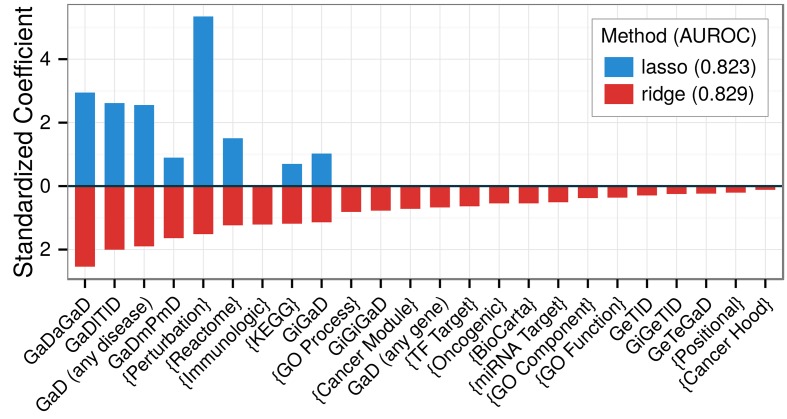
The first decade of Genome Wide Association Studies (GWAS) has uncovered a wealth of disease-associated variants. Two important derivations will be the translation of this information into a multiscale understanding of pathogenic variants and leveraging existing data to increase the power of existing and future studies through prioritization. We explore edge prediction on heterogeneous networks--graphs with multiple node and edge types--for accomplishing both tasks. First we constructed a network with 18 node types--genes, diseases, tissues, pathophysiologies, and 14 MSigDB (molecular signatures database) collections--and 19 edge types from high-throughput publicly-available resources. From this network composed of 40,343 nodes and 1,608,168 edges, we extracted features that describe the topology between specific genes and diseases. Next, we trained a model from GWAS associations and predicted the probability of association between each protein-coding gene and each of 29 well-studied complex diseases. The model, which achieved 132-fold enrichment in precision at 10% recall, outperformed any individual domain, highlighting the benefit of integrative approaches. We identified pleiotropy, transcriptional signatures of perturbations, pathways, and protein interactions as influential mechanisms explaining pathogenesis. Our method successfully predicted the results (with AUROC = 0.79) from a withheld multiple sclerosis (MS) GWAS despite starting with only 13 previously associated genes. Finally, we combined our network predictions with statistical evidence of association to propose four novel MS genes, three of which (JAK2, REL, RUNX3) validated on the masked GWAS. Furthermore, our predictions provide biological support highlighting REL as the causal gene within its gene-rich locus. Users can browse all predictions online (http://het.io). Heterogeneous network edge prediction effectively prioritized genetic associations and provides a powerful new approach for data integration across multiple domains.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
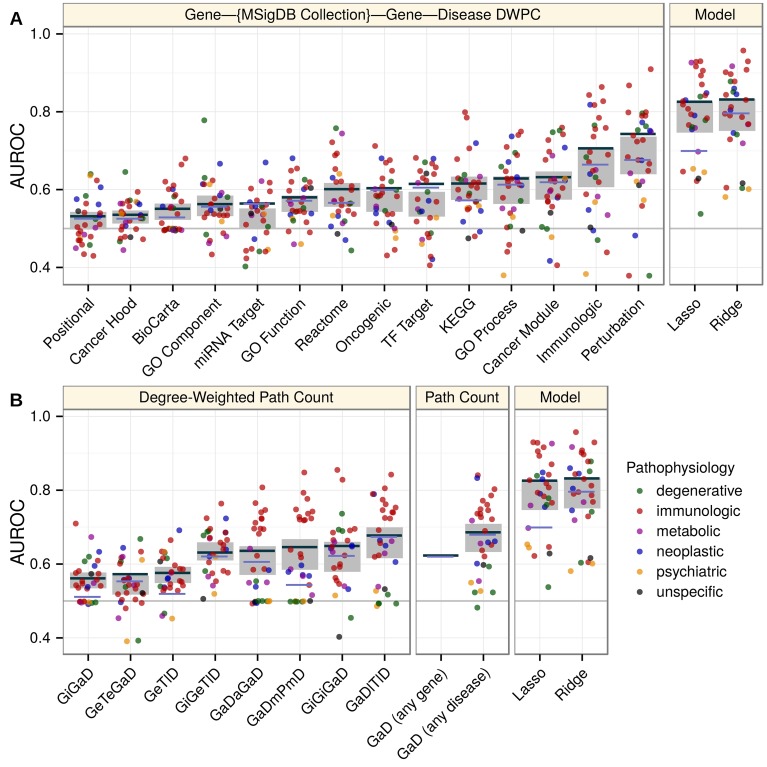
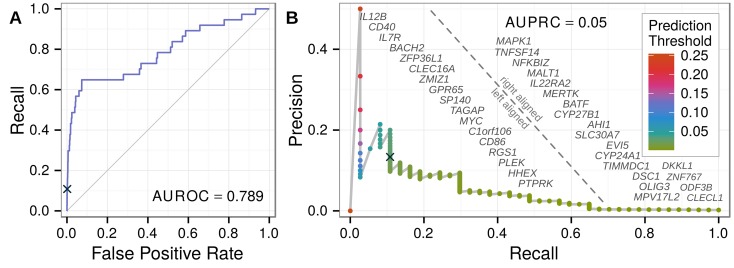
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
