

Lansoprazole is an antituberculous prodrug targeting cytochrome bc1
- PMID: 26158909
- PMCID: PMC4510652
- DOI: 10.1038/ncomms8659
Lansoprazole is an antituberculous prodrug targeting cytochrome bc1
Abstract
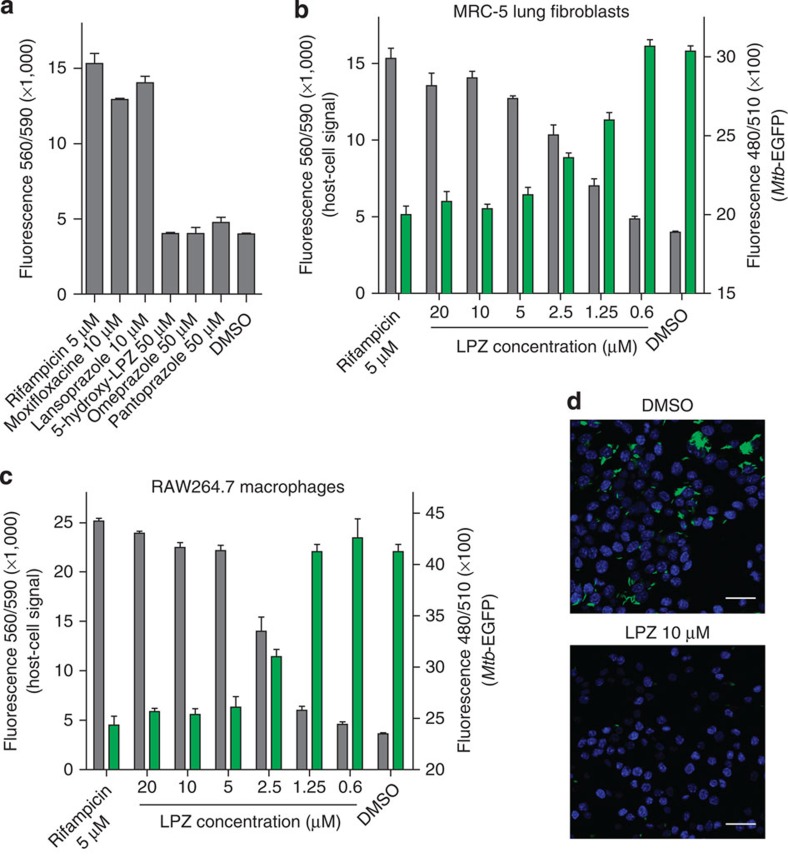
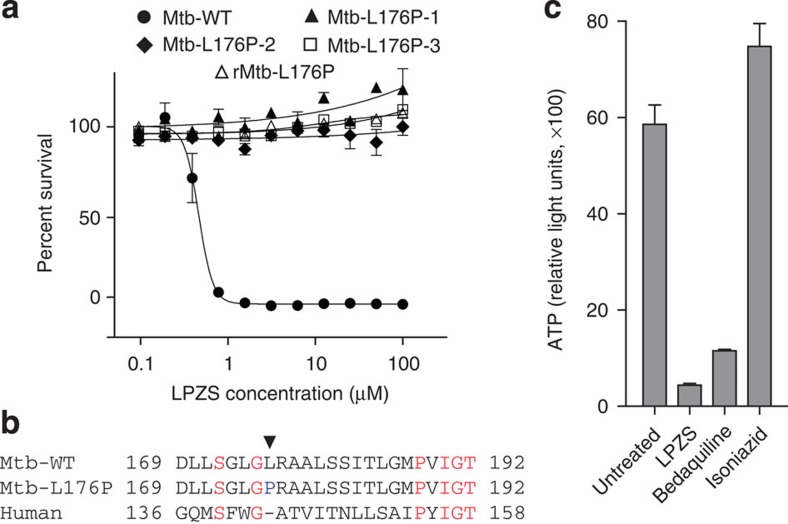
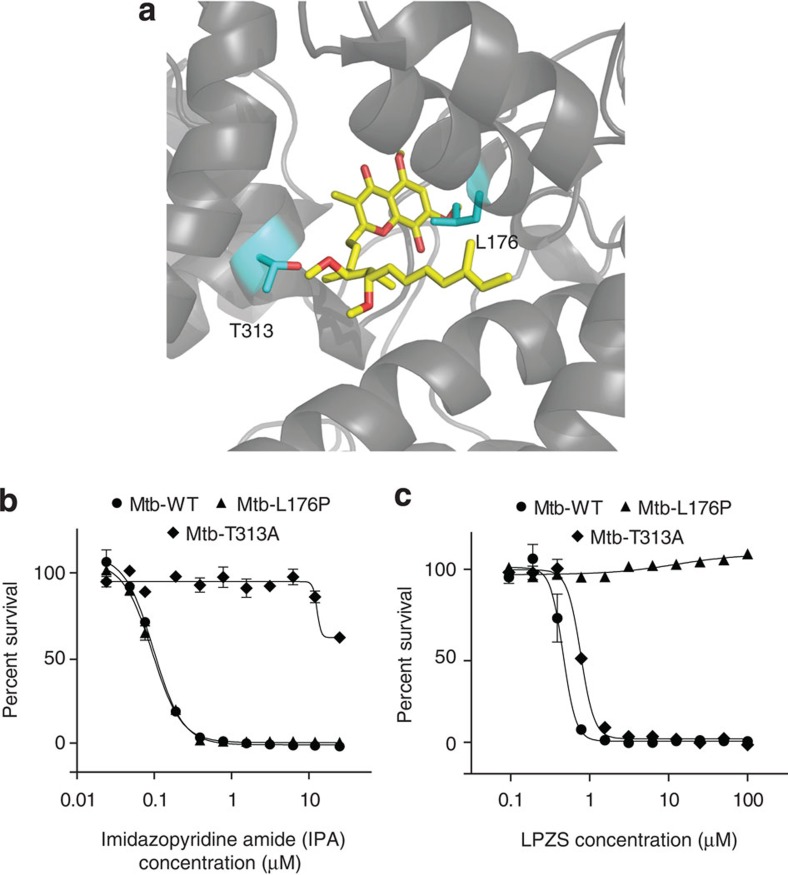
Better antibiotics capable of killing multi-drug-resistant Mycobacterium tuberculosis are urgently needed. Despite extensive drug discovery efforts, only a few promising candidates are on the horizon and alternative screening protocols are required. Here, by testing a panel of FDA-approved drugs in a host cell-based assay, we show that the blockbuster drug lansoprazole (Prevacid), a gastric proton-pump inhibitor, has intracellular activity against M. tuberculosis. Ex vivo pharmacokinetics and target identification studies reveal that lansoprazole kills M. tuberculosis by targeting its cytochrome bc1 complex through intracellular sulfoxide reduction to lansoprazole sulfide. This novel class of cytochrome bc1 inhibitors is highly active against drug-resistant clinical isolates and spares the human H(+)K(+)-ATPase thus providing excellent opportunities for targeting the major pathogen M. tuberculosis. Our finding provides proof of concept for hit expansion by metabolic activation, a powerful tool for antibiotic screens.
Conflict of interest statement
J.R. and S.T.C. are named inventors on a patent pertaining to this work. The remaining authors declare no competing financial interests.
Figures


References
-
- Lewis K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 12, 371–387 (2013). - PubMed
-
- World Health Organization. Global Tuberculosis Report 2014 WHO (2014) http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf?....
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
