

MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases
- PMID: 26159306
- PMCID: PMC4817924
- DOI: 10.1038/mt.2015.126
MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases
Abstract
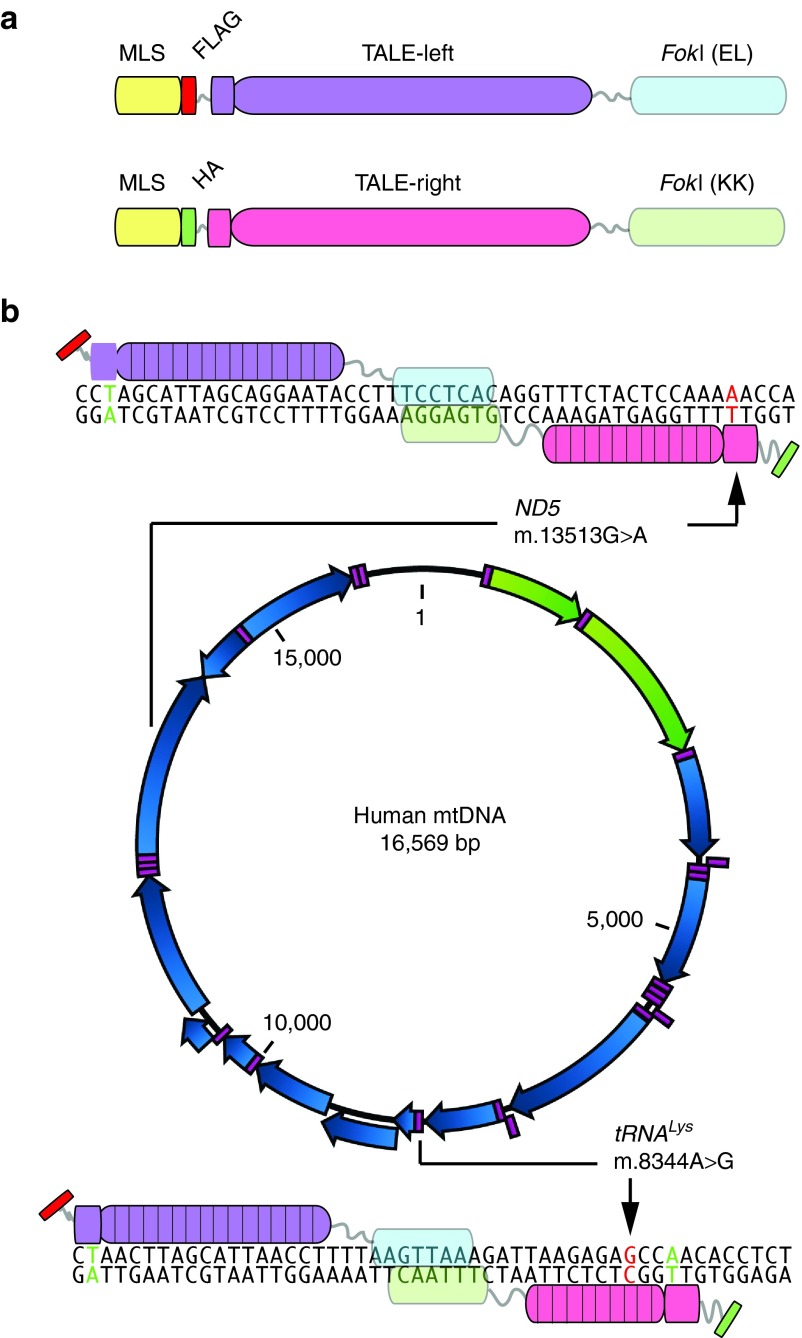
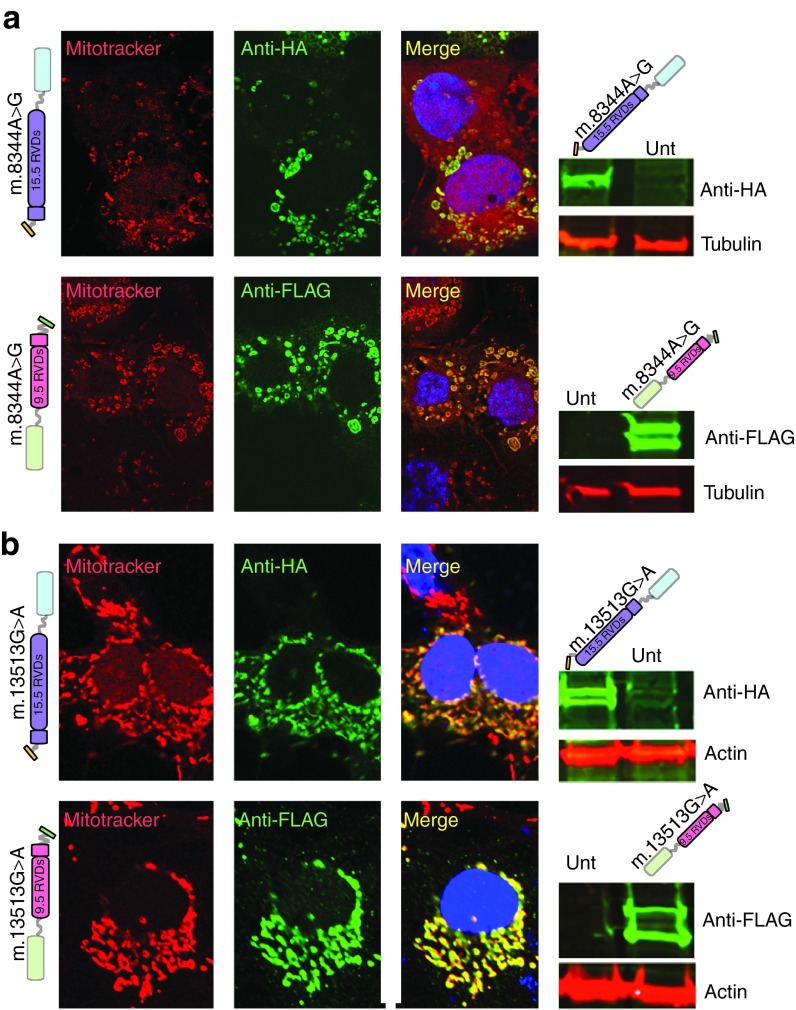
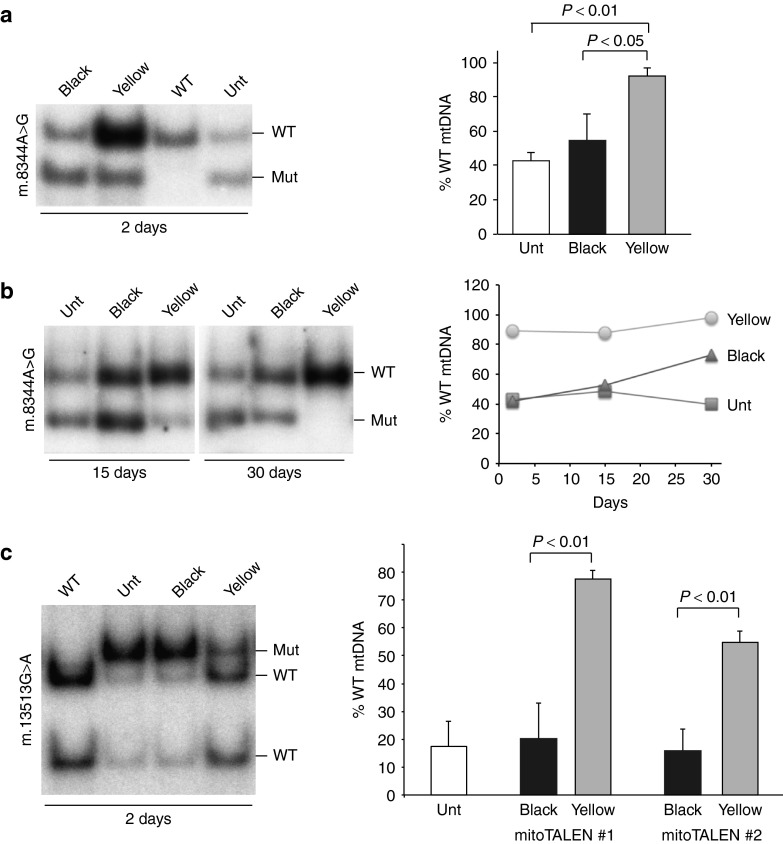
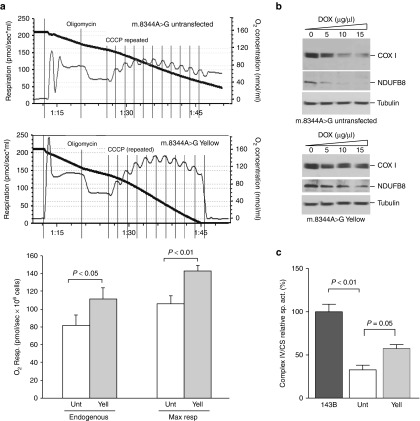
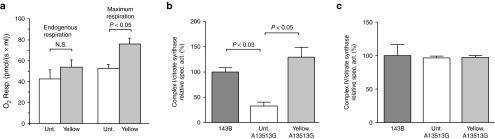
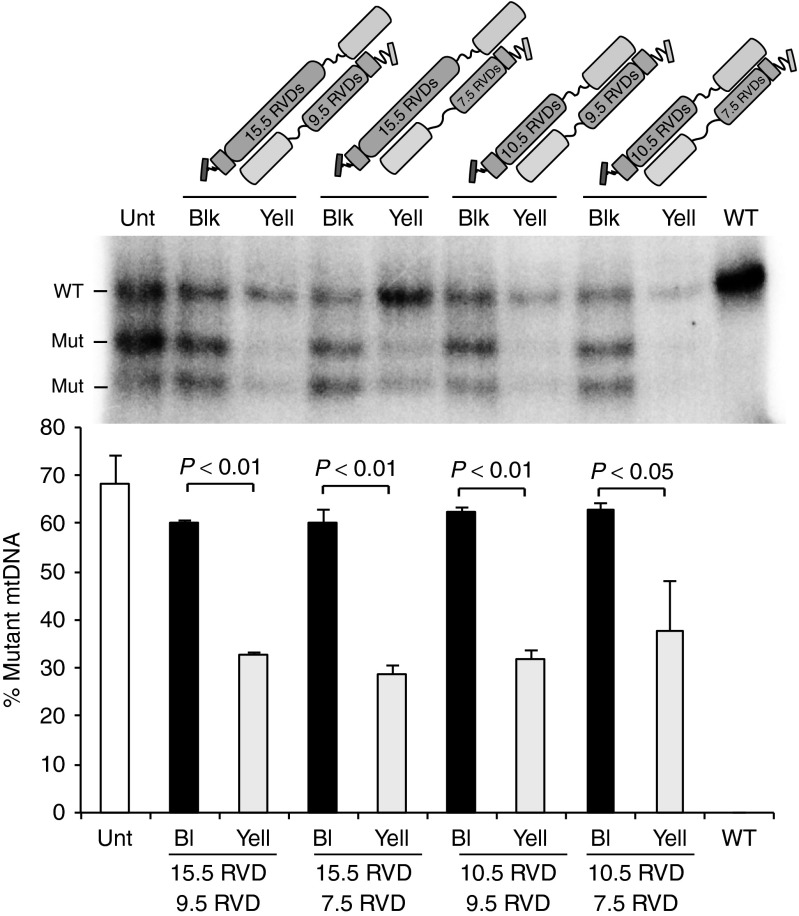
We have designed mitochondrially targeted transcription activator-like effector nucleases or mitoTALENs to cleave specific sequences in the mitochondrial DNA (mtDNA) with the goal of eliminating mtDNA carrying pathogenic point mutations. To test the generality of the approach, we designed mitoTALENs to target two relatively common pathogenic mtDNA point mutations associated with mitochondrial diseases: the m.8344A>G tRNA(Lys) gene mutation associated with myoclonic epilepsy with ragged red fibers (MERRF) and the m.13513G>A ND5 mutation associated with MELAS/Leigh syndrome. Transmitochondrial cybrid cells harbouring the respective heteroplasmic mtDNA mutations were transfected with the respective mitoTALEN and analyzed after different time periods. MitoTALENs efficiently reduced the levels of the targeted pathogenic mtDNAs in the respective cell lines. Functional assays showed that cells with heteroplasmic mutant mtDNA were able to recover respiratory capacity and oxidative phosphorylation enzymes activity after transfection with the mitoTALEN. To improve the design in the context of the low complexity of mtDNA, we designed shorter versions of the mitoTALEN specific for the MERRF m.8344A>G mutation. These shorter mitoTALENs also eliminated the mutant mtDNA. These reductions in size will improve our ability to package these large sequences into viral vectors, bringing the use of these genetic tools closer to clinical trials.
Figures
Similar articles
-
Mutations in the ND5 subunit of complex I of the mitochondrial DNA are a frequent cause of oxidative phosphorylation disease.J Med Genet. 2007 Apr;44(4):e74. doi: 10.1136/jmg.2006.045716. J Med Genet. 2007. PMID: 17400793 Free PMC article.
-
MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation.Nat Med. 2018 Nov;24(11):1696-1700. doi: 10.1038/s41591-018-0166-8. Epub 2018 Sep 24. Nat Med. 2018. PMID: 30250143 Free PMC article.
-
Modeling of antigenomic therapy of mitochondrial diseases by mitochondrially addressed RNA targeting a pathogenic point mutation in mitochondrial DNA.J Biol Chem. 2014 May 9;289(19):13323-34. doi: 10.1074/jbc.M113.528968. Epub 2014 Apr 1. J Biol Chem. 2014. PMID: 24692550 Free PMC article.
-
Modulating Mitochondrial DNA Heteroplasmy with Mitochondrially Targeted Endonucleases.Ann Biomed Eng. 2024 Sep;52(9):2627-2640. doi: 10.1007/s10439-022-03051-7. Epub 2022 Aug 24. Ann Biomed Eng. 2024. PMID: 36001180 Free PMC article. Review.
-
Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches.EMBO Rep. 2020 Mar 4;21(3):e49612. doi: 10.15252/embr.201949612. Epub 2020 Feb 19. EMBO Rep. 2020. PMID: 32073748 Free PMC article. Review.
Cited by
-
Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome.Essays Biochem. 2018 Jul 20;62(3):455-465. doi: 10.1042/EBC20170113. Print 2018 Jul 20. Essays Biochem. 2018. PMID: 29950320 Free PMC article. Review.
-
Induce male sterility by CRISPR/Cas9-mediated mitochondrial genome editing in tobacco.Funct Integr Genomics. 2023 Jun 19;23(3):205. doi: 10.1007/s10142-023-01136-7. Funct Integr Genomics. 2023. PMID: 37335501
-
A novel correlation between ATP5A1 gene expression and progression of human clear cell renal cell carcinoma identified by co‑expression analysis.Oncol Rep. 2018 Feb;39(2):525-536. doi: 10.3892/or.2017.6132. Epub 2017 Dec 4. Oncol Rep. 2018. PMID: 29207195 Free PMC article.
-
Elimination of Mutant mtDNA by an Optimized mpTALEN Restores Differentiation Capacities of Heteroplasmic MELAS-iPSCs.Mol Ther Methods Clin Dev. 2020 Oct 22;20:54-68. doi: 10.1016/j.omtm.2020.10.017. eCollection 2021 Mar 12. Mol Ther Methods Clin Dev. 2020. PMID: 33376755 Free PMC article.
-
Generation and Bioenergetic Profiles of Cybrids with East Asian mtDNA Haplogroups.Oxid Med Cell Longev. 2017;2017:1062314. doi: 10.1155/2017/1062314. Epub 2017 Sep 28. Oxid Med Cell Longev. 2017. PMID: 29093766 Free PMC article.
References
-
- Thorburn, DR and Dahl, HH (2001). Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. Am J Med Genet 106: 102–114. - PubMed
-
- DiMauro, S and Schon, EA (2003). Mitochondrial respiratory-chain diseases. N Engl J Med 348: 2656–2668. - PubMed
-
- Gardner, JL, Craven, L, Turnbull, DM and Taylor, RW (2007). Experimental strategies towards treating mitochondrial DNA disorders. Biosci Rep 27: 139–150. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
