

EXO1 is critical for embryogenesis and the DNA damage response in mice with a hypomorphic Nbs1 allele
- PMID: 26160886
- PMCID: PMC4551929
- DOI: 10.1093/nar/gkv691
EXO1 is critical for embryogenesis and the DNA damage response in mice with a hypomorphic Nbs1 allele
Abstract
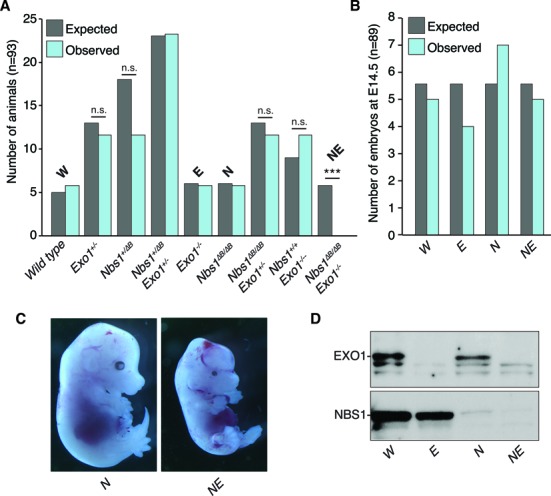
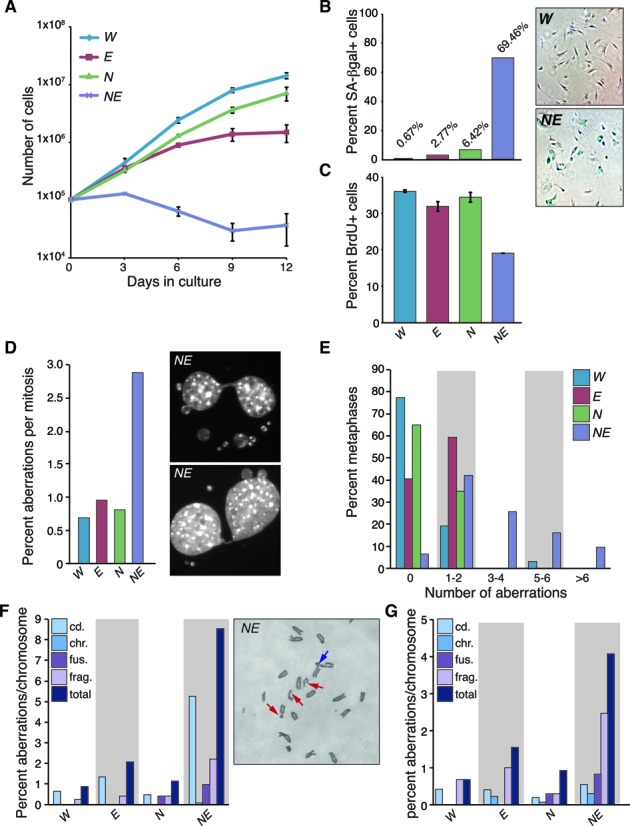
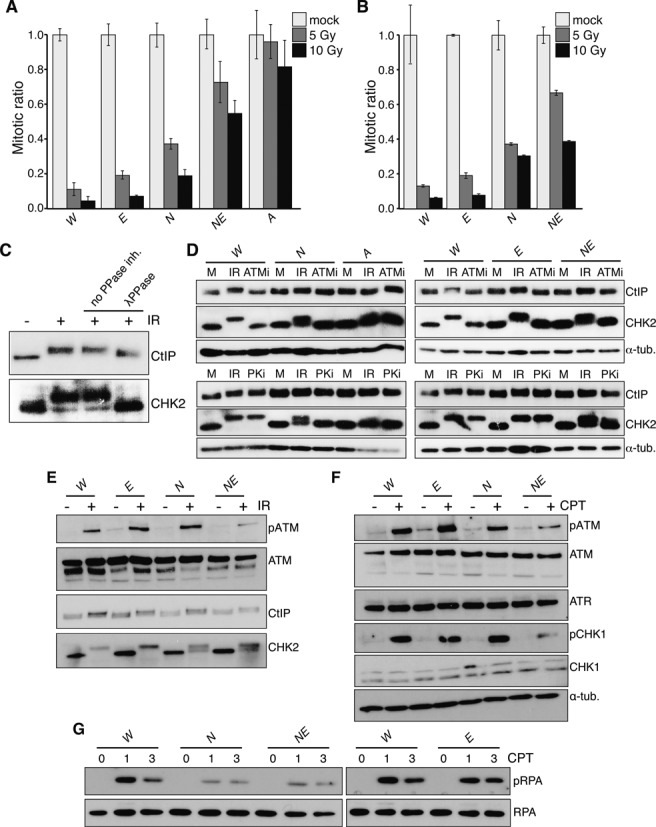
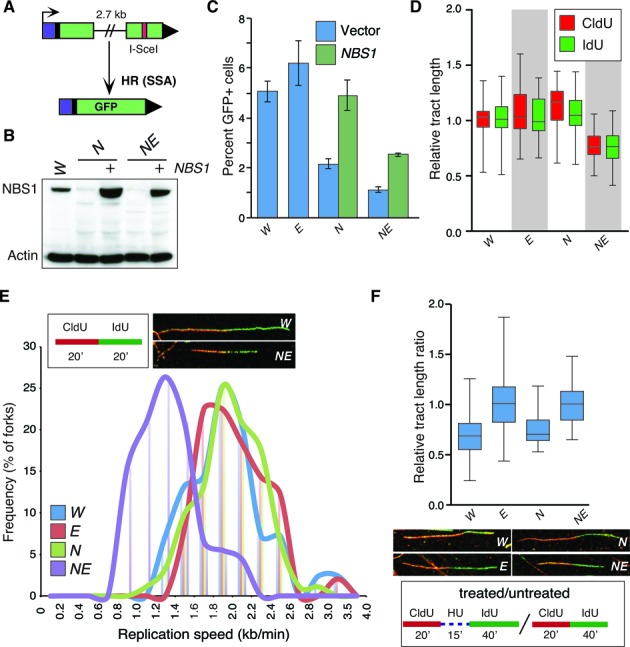
The maintenance of genome stability is critical for the suppression of diverse human pathologies that include developmental disorders, premature aging, infertility and predisposition to cancer. The DNA damage response (DDR) orchestrates the appropriate cellular responses following the detection of lesions to prevent genomic instability. The MRE11 complex is a sensor of DNA double strand breaks (DSBs) and plays key roles in multiple aspects of the DDR, including DNA end resection that is critical for signaling and DNA repair. The MRE11 complex has been shown to function both upstream and in concert with the 5'-3' exonuclease EXO1 in DNA resection, but it remains unclear to what extent EXO1 influences DSB responses independently of the MRE11 complex. Here we examine the genetic relationship of the MRE11 complex and EXO1 during mammalian development and in response to DNA damage. Deletion of Exo1 in mice expressing a hypomorphic allele of Nbs1 leads to severe developmental impairment, embryonic death and chromosomal instability. While EXO1 plays a minimal role in normal cells, its loss strongly influences DNA replication, DNA repair, checkpoint signaling and damage sensitivity in NBS1 hypomorphic cells. Collectively, our results establish a key role for EXO1 in modulating the severity of hypomorphic MRE11 complex mutations.
© The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
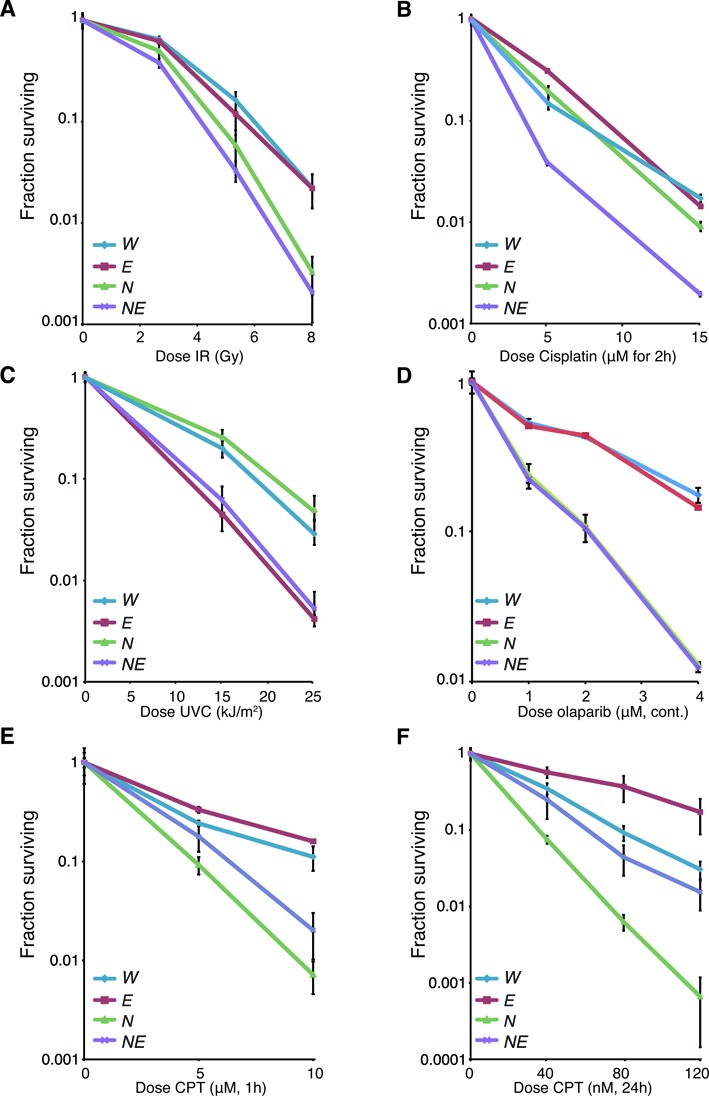
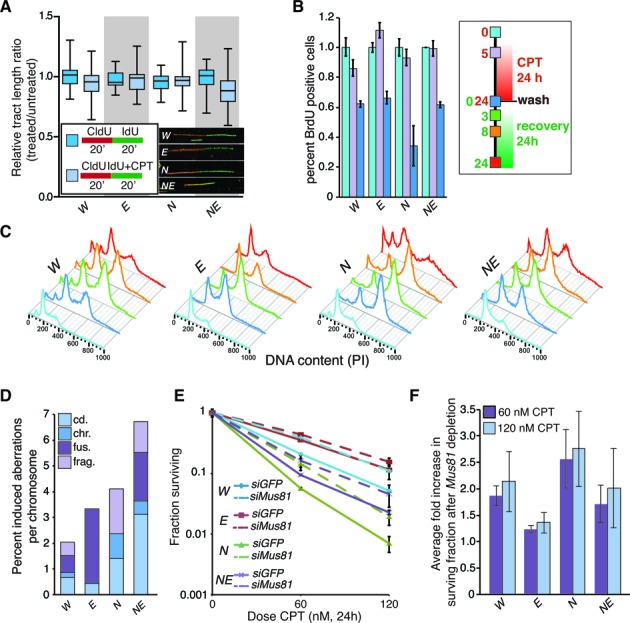
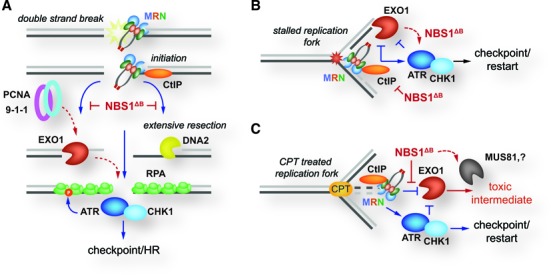
References
-
- Halazonetis T.D., Gorgoulis V.G., Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
