

Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome
- PMID: 26166481
- PMCID: PMC4573270
- DOI: 10.1016/j.ajhg.2015.06.003
Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome
Abstract
KIAA0586, the human ortholog of chicken TALPID3, is a centrosomal protein that is essential for primary ciliogenesis. Its disruption in animal models causes defects attributed to abnormal hedgehog signaling; these defects include polydactyly and abnormal dorsoventral patterning of the neural tube. Here, we report homozygous mutations of KIAA0586 in four families affected by lethal ciliopathies ranging from a hydrolethalus phenotype to short-rib polydactyly. We show defective ciliogenesis, as well as abnormal response to SHH-signaling activation in cells derived from affected individuals, consistent with a role of KIAA0586 in primary cilia biogenesis. Whereas centriolar maturation seemed unaffected in mutant cells, we observed an abnormal extended pattern of CEP290, a centriolar satellite protein previously associated with ciliopathies. Our data show the crucial role of KIAA0586 in human primary ciliogenesis and subsequent abnormal hedgehog signaling through abnormal GLI3 processing. Our results thus establish that KIAA0586 mutations cause lethal ciliopathies.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
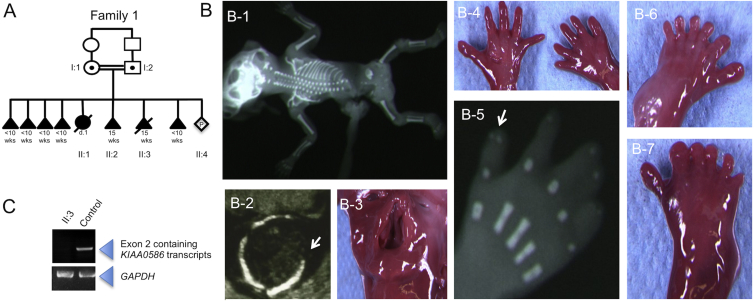
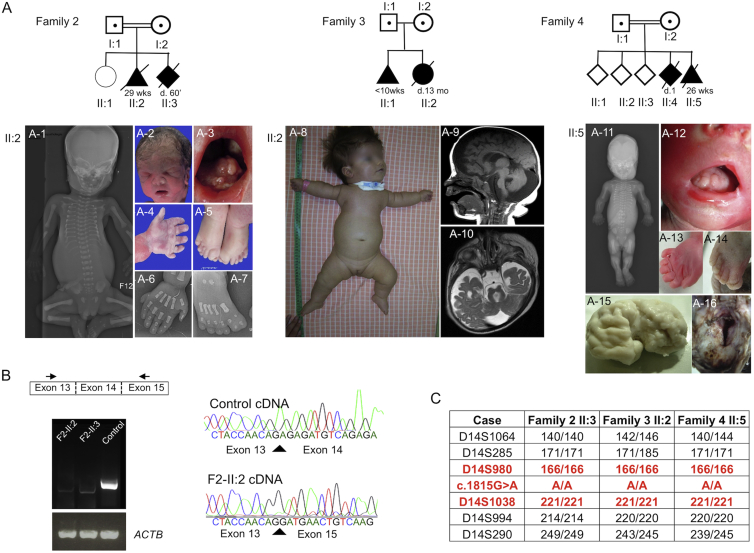
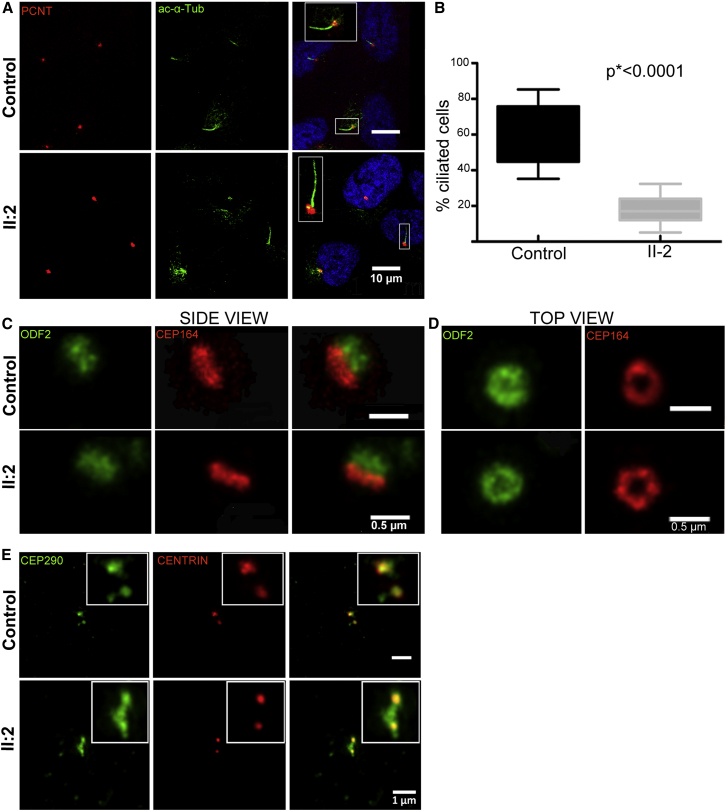
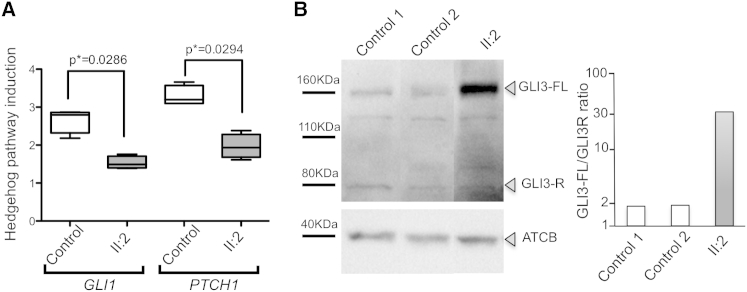
References
-
- Huber C., Cormier-Daire V. Ciliary disorder of the skeleton. Am. J. Med. Genet. C. Semin. Med. Genet. 2012;160C:165–174. - PubMed
-
- Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
