

An integrative pan-cancer-wide analysis of epigenetic enzymes reveals universal patterns of epigenomic deregulation in cancer
- PMID: 26169266
- PMCID: PMC4501092
- DOI: 10.1186/s13059-015-0699-9
An integrative pan-cancer-wide analysis of epigenetic enzymes reveals universal patterns of epigenomic deregulation in cancer
Abstract
Background: One of the most important recent findings in cancer genomics is the identification of novel driver mutations which often target genes that regulate genome-wide chromatin and DNA methylation marks. Little is known, however, as to whether these genes exhibit patterns of epigenomic deregulation that transcend cancer types.
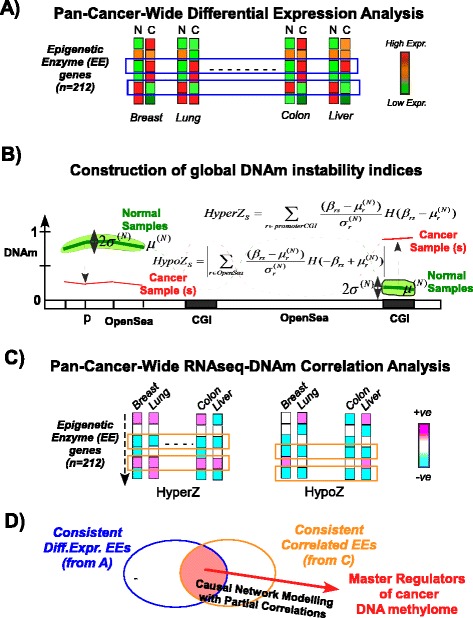
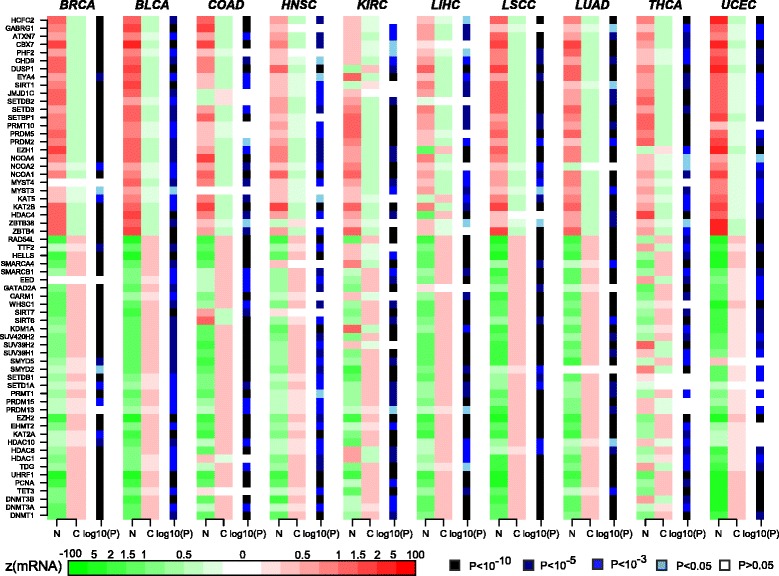
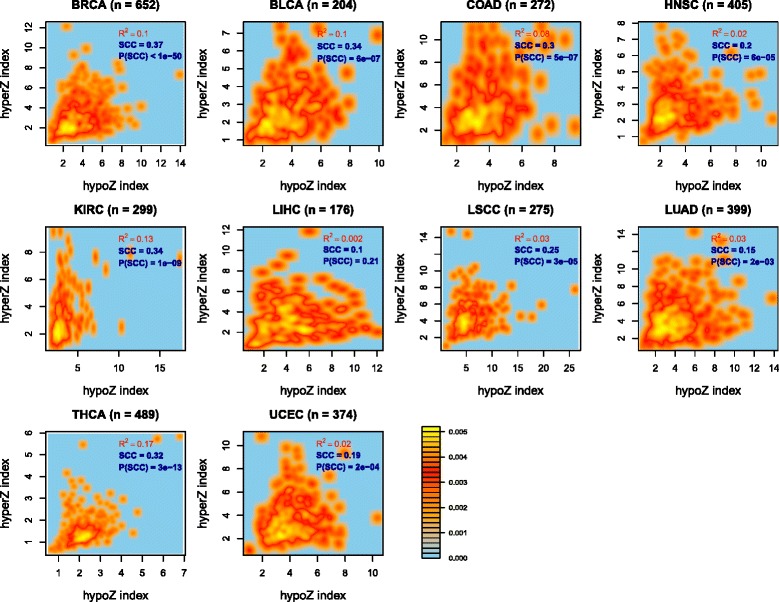
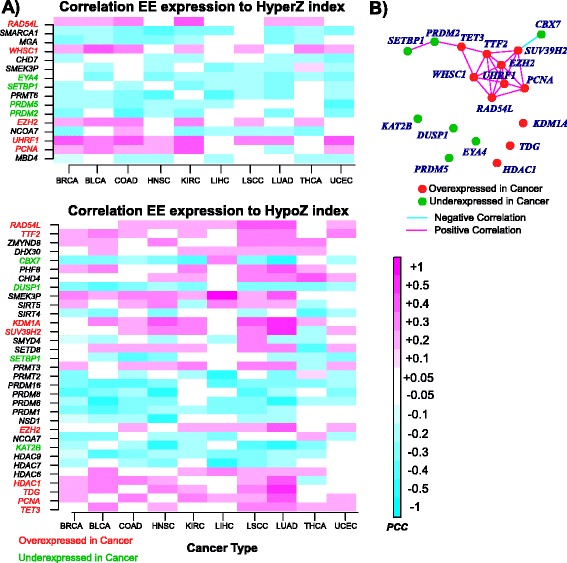
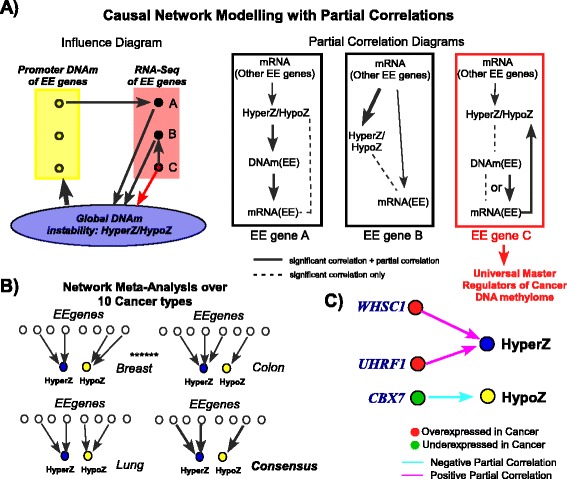
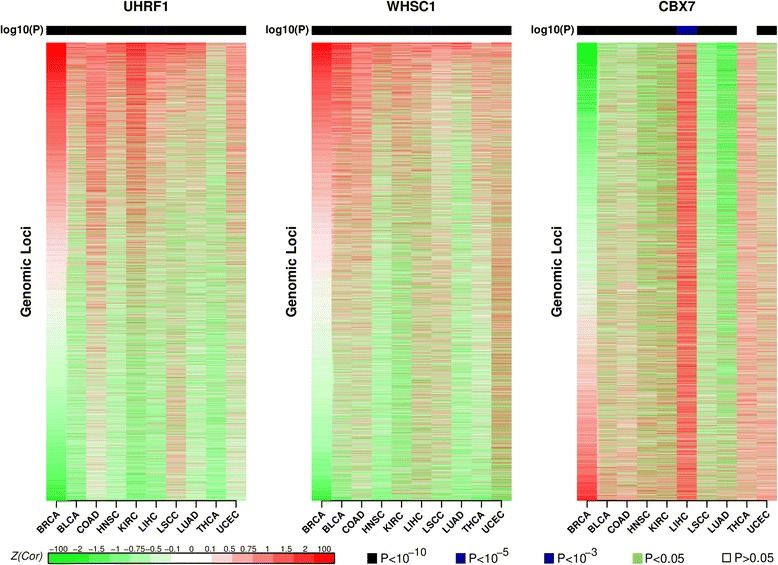
Results: Here we conduct an integrative pan-cancer-wide analysis of matched RNA-Seq and DNA methylation data across ten different cancer types. We identify seven tumor suppressor and eleven oncogenic epigenetic enzymes which display patterns of deregulation and association with genome-wide cancer DNA methylation patterns, which are largely independent of cancer type. In doing so, we provide evidence that genome-wide cancer hyper- and hypo- DNA methylation patterns are independent processes, controlled by distinct sets of epigenetic enzyme genes. Using causal network modeling, we predict a number of candidate drivers of cancer DNA hypermethylation and hypomethylation. Finally, we show that the genomic loci whose DNA methylation levels associate most strongly with expression of these putative drivers are highly consistent across cancer types.
Conclusions: This study demonstrates that there exist universal patterns of epigenomic deregulation that transcend cancer types, and that intra-tumor levels of genome-wide DNA hypomethylation and hypermethylation are controlled by distinct processes.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
