

A 13-year follow-up of Finnish patients with Salla disease
- PMID: 26171070
- PMCID: PMC4499899
- DOI: 10.1186/s11689-015-9116-7
A 13-year follow-up of Finnish patients with Salla disease
Abstract
Background: Salla disease (SD) is a rare lysosomal storage disorder leading to severe intellectual disability. SD belongs to the Finnish disease heritage, and it is caused by mutations in the SLC17A5 gene. The aim of the study was to investigate the course of neurocognitive features of SD patients in a long-term follow-up.
Methods: Neuropsychological and neurological investigations were carried out on 24 SD patients, aged 16-65 years, 13 years after a similar examination.
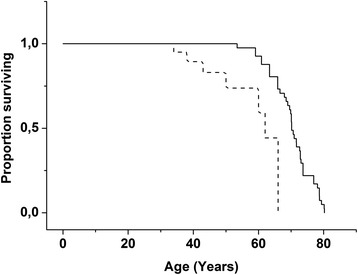
Results: The survival analysis showed excess mortality among patients with SD after the age of 30 years. The course of the disease was progressive, but follow-up of SD patients revealed that motor skills improved till the age of 20 years, while mental abilities improved in most patients till 40 years of age. Verbal comprehension skills did not diminish during the follow-up, but productive speech deteriorated because of dyspraxia and dysarthria. Motor deficits were marked. Ataxia was prominent in childhood, but it was replaced by athetotic movements during the teens. Spasticity became more obvious with age especially in severely disabled SD patients.
Conclusions: Younger SD patients performed better in almost every task measuring mental abilities that then seem to remain fairly constant till early sixties. Thus, the results indicate better prognosis in cognitive skills than earlier assumed. There is an apparent decline in motor skills after the age of 20 years. The early neurocognitive development predicts the later course of motor and cognitive development.
Keywords: Dysmyelination; Follow-up study; Free sialic acid storage; Neurocognitive development; Rare diseases.
Figures


Similar articles
-
Neurocognitive profiles in Salla disease.Dev Med Child Neurol. 2004 Dec;46(12):832-7. doi: 10.1017/s0012162204001458. Dev Med Child Neurol. 2004. PMID: 15581157
-
Phenotypic spectrum of Salla disease, a free sialic acid storage disorder.Pediatr Neurol. 2002 Apr;26(4):267-73. doi: 10.1016/s0887-8994(01)00406-4. Pediatr Neurol. 2002. PMID: 11992753
-
An Italian severe Salla disease variant associated with a SLC17A5 mutation earlier described in infantile sialic acid storage disease.Clin Genet. 2002 Jun;61(6):443-7. doi: 10.1034/j.1399-0004.2002.610608.x. Clin Genet. 2002. PMID: 12121352
-
A New Patient With Intermediate Severe Salla Disease With Hypomyelination: A Literature Review for Salla Disease.Pediatr Neurol. 2017 Sep;74:87-91.e2. doi: 10.1016/j.pediatrneurol.2017.05.022. Epub 2017 Jun 1. Pediatr Neurol. 2017. PMID: 28662915 Review.
-
[Lysosomal membrane transport disorders--cystinosis and sialic acid storage disorders (Salla disease, ISSD)].Nihon Rinsho. 1995 Dec;53(12):3068-71. Nihon Rinsho. 1995. PMID: 8577060 Review. Japanese.
Cited by
-
Base editing corrects the common Salla disease SLC17A5 c.115C>T variant.Mol Ther Nucleic Acids. 2023 Aug 26;34:102022. doi: 10.1016/j.omtn.2023.08.024. eCollection 2023 Dec 12. Mol Ther Nucleic Acids. 2023. PMID: 37727271 Free PMC article.
-
Psychiatric symptoms in Salla disease.Eur Child Adolesc Psychiatry. 2023 Oct;32(10):2043-2047. doi: 10.1007/s00787-022-02031-5. Epub 2022 Jul 7. Eur Child Adolesc Psychiatry. 2023. PMID: 35796883 Free PMC article. Review.
References
-
- Norio R. The Finnish disease heritage III: the individual diseases. Hum Genet. 2003;112(5-6):470–526. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
