

JAK/STAT pathway regulation of GABAA receptor expression after differing severities of experimental TBI
- PMID: 26172316
- PMCID: PMC5969808
- DOI: 10.1016/j.expneurol.2015.07.001
JAK/STAT pathway regulation of GABAA receptor expression after differing severities of experimental TBI
Abstract
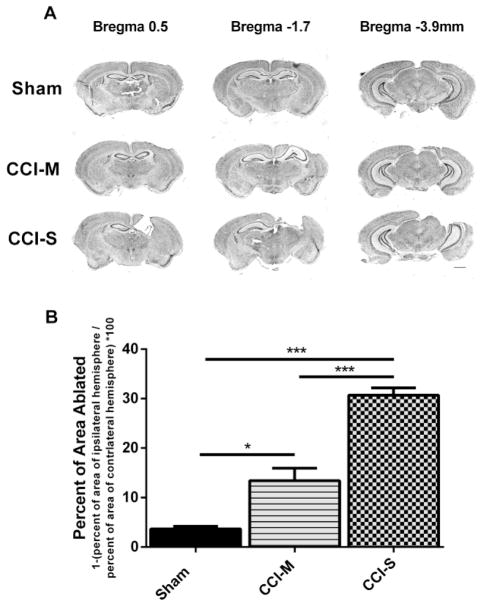
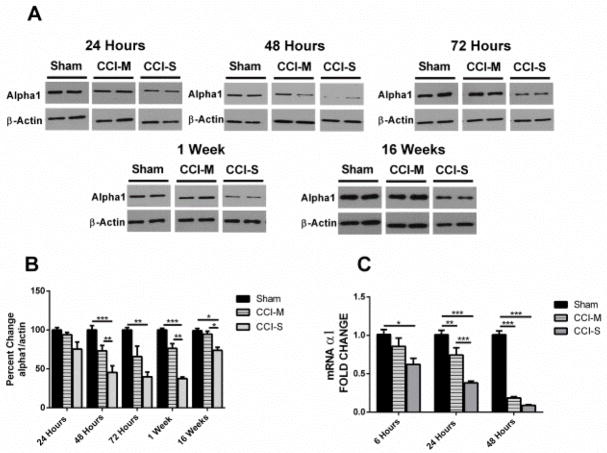
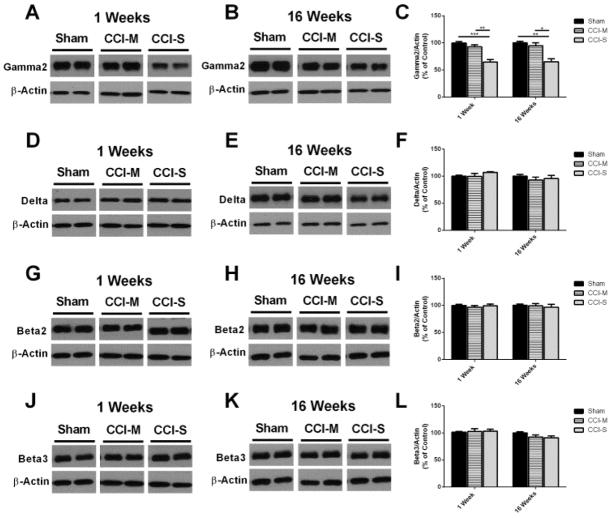
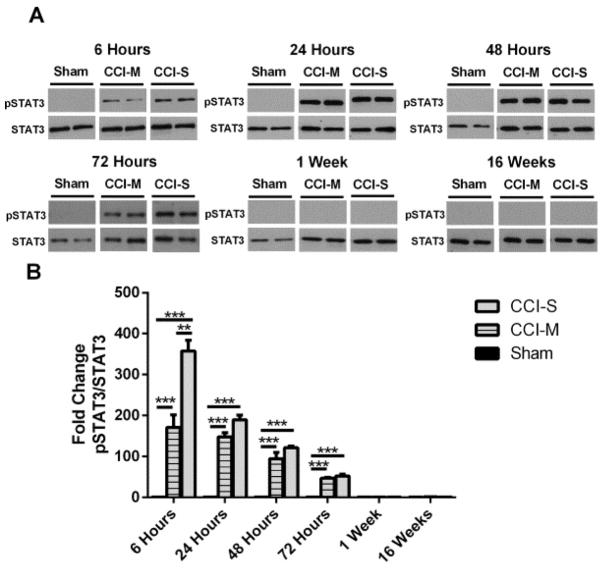
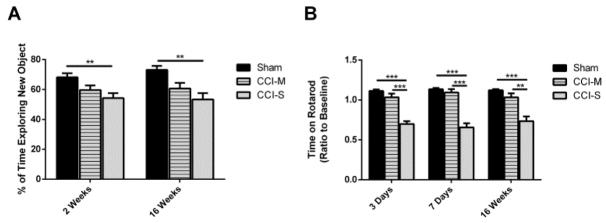
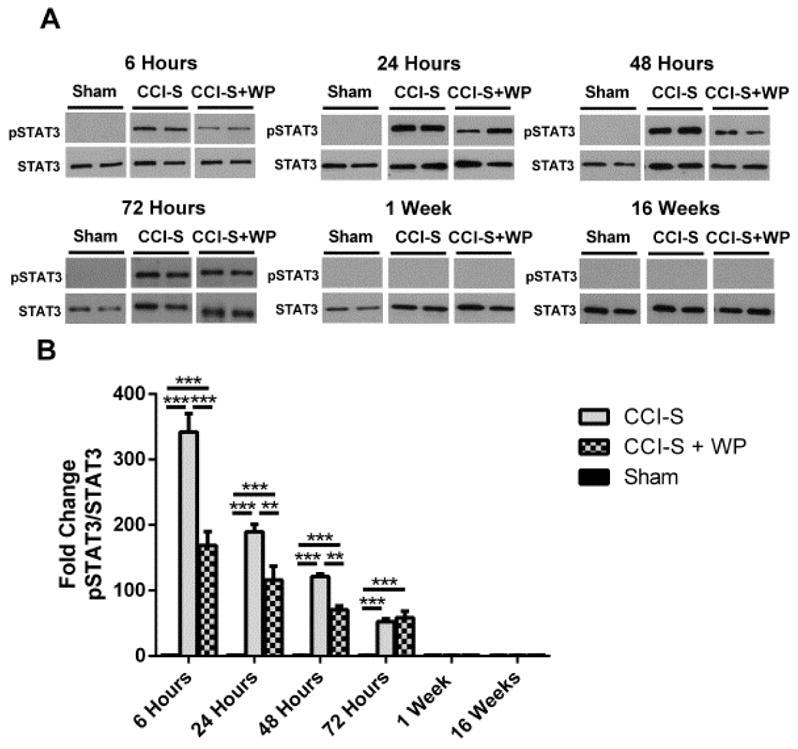
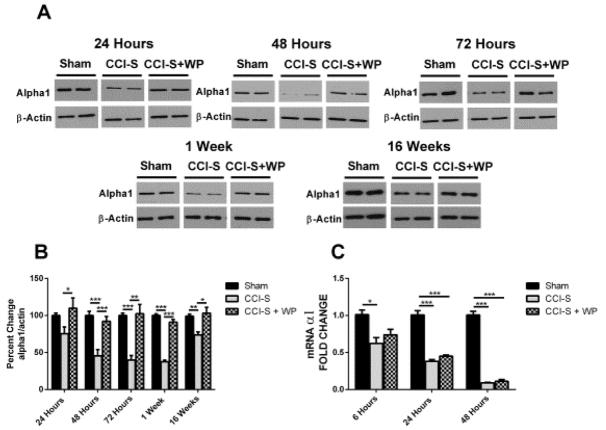
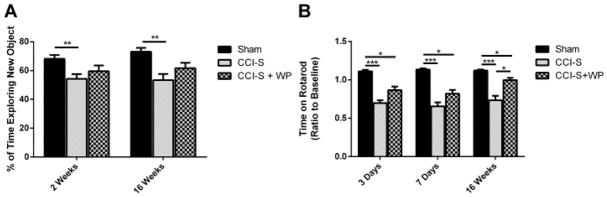
Synaptic inhibition in the adult brain is primarily mediated by the γ-aminobutyric acid (GABA) type A receptor (GABA(A)R). The distribution, properties, and dynamics of these receptors are largely determined by their subunit composition. Alteration of subunit composition after a traumatic brain injury (TBI) may result in abnormal increased synaptic firing and possibly contribute to injury-related pathology. Several studies have shown that the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling pathway can alter GABA(A)R subunit expression. The present study investigated changes in JAK/STAT pathway activation after two different severities of experimental TBI in the mouse using the controlled cortical impact (CCI) model. It also investigated whether modulating the activation of the JAK/STAT pathway after severe controlled cortical impact (CCI-S) with a JAK/STAT inhibitor (WP1066) alters post-traumatic epilepsy development and/or neurological recovery after injury. Our results demonstrated differential changes in both the activation of STAT3 and the expression of the GABA(A)R α1 and γ2 subunit levels that were dependent on the severity of the injury. The change in the GABA(A)R α1 subunit levels appeared to be at least partly transcriptionally mediated. We were able to selectively reverse the decrease in GABA(A)R α1 protein levels with WP1066 treatment after CCI injury. WP1066 treatment also improved the degree of recovery of vestibular motor function after injury. These findings suggest that the magnitude of JAK/STAT pathway activation and GABA(A)R α1 subunit level decrease is dependent on injury severity in this mouse model of TBI. In addition, reducing JAK/STAT pathway activation after severe experimental TBI reverses the decrease in the GABA(A)R α1 protein levels and improves vestibular motor recovery.
Keywords: GABA(A) receptor; JAK/STAT pathway; STAT3; Traumatic brain injury.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
GABA(A) receptor regulation after experimental traumatic brain injury.J Neurotrauma. 2012 Nov 1;29(16):2548-54. doi: 10.1089/neu.2012.2483. Epub 2012 Sep 4. J Neurotrauma. 2012. PMID: 22827467 Free PMC article.
-
BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway.Sci Signal. 2008 Oct 14;1(41):ra9. doi: 10.1126/scisignal.1162396. Sci Signal. 2008. PMID: 18922788 Free PMC article.
-
Canonical JAK-STAT signaling is pivotal for long-term depression at adult hippocampal temporoammonic-CA1 synapses.FASEB J. 2017 Aug;31(8):3449-3466. doi: 10.1096/fj.201601293RR. Epub 2017 May 1. FASEB J. 2017. PMID: 28461339
-
The JAK-STAT Signaling Pathway in Epilepsy.Curr Neuropharmacol. 2023;21(10):2049-2069. doi: 10.2174/1570159X21666221214170234. Curr Neuropharmacol. 2023. PMID: 36518035 Free PMC article. Review.
-
The JAK/STAT signaling pathway: from bench to clinic.Signal Transduct Target Ther. 2021 Nov 26;6(1):402. doi: 10.1038/s41392-021-00791-1. Signal Transduct Target Ther. 2021. PMID: 34824210 Free PMC article. Review.
Cited by
-
Selective vulnerability of hippocampal interneurons to graded traumatic brain injury.Neurobiol Dis. 2019 Sep;129:208-216. doi: 10.1016/j.nbd.2018.07.022. Epub 2018 Jul 19. Neurobiol Dis. 2019. PMID: 30031783 Free PMC article. Review.
-
Zolpidem Profoundly Augments Spared Tonic GABAAR Signaling in Dentate Granule Cells Ipsilateral to Controlled Cortical Impact Brain Injury in Mice.Front Syst Neurosci. 2022 May 26;16:867323. doi: 10.3389/fnsys.2022.867323. eCollection 2022. Front Syst Neurosci. 2022. PMID: 35694044 Free PMC article.
-
Janus kinase inhibitors are potential therapeutics for amyotrophic lateral sclerosis.Transl Neurodegener. 2023 Oct 12;12(1):47. doi: 10.1186/s40035-023-00380-y. Transl Neurodegener. 2023. PMID: 37828541 Free PMC article. Review.
-
Cellular and Molecular Pathophysiology of Traumatic Brain Injury: What Have We Learned So Far?Biology (Basel). 2023 Aug 17;12(8):1139. doi: 10.3390/biology12081139. Biology (Basel). 2023. PMID: 37627023 Free PMC article. Review.
-
Alcohol and IL-6 Alter Expression of Synaptic Proteins in Cerebellum of Transgenic Mice with Increased Astrocyte Expression of IL-6.Neuroscience. 2020 Aug 21;442:124-137. doi: 10.1016/j.neuroscience.2020.06.043. Epub 2020 Jul 4. Neuroscience. 2020. PMID: 32634532 Free PMC article.
References
-
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. The New England journal of medicine. 1998;338:20–24. - PubMed
-
- Bolkvadze T, Pitkanen A. Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. Journal of neurotrauma. 2012;29:789–812. - PubMed
-
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. - PubMed
-
- Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke; a journal of cerebral circulation. 2001;32:1005–1011. - PubMed
-
- Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E. An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. Journal of neurotrauma. 1996;13:557–568. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
