

Lysophosphatidic Acid Initiates Epithelial to Mesenchymal Transition and Induces β-Catenin-mediated Transcription in Epithelial Ovarian Carcinoma
- PMID: 26175151
- PMCID: PMC4571965
- DOI: 10.1074/jbc.M115.641092
Lysophosphatidic Acid Initiates Epithelial to Mesenchymal Transition and Induces β-Catenin-mediated Transcription in Epithelial Ovarian Carcinoma
Abstract
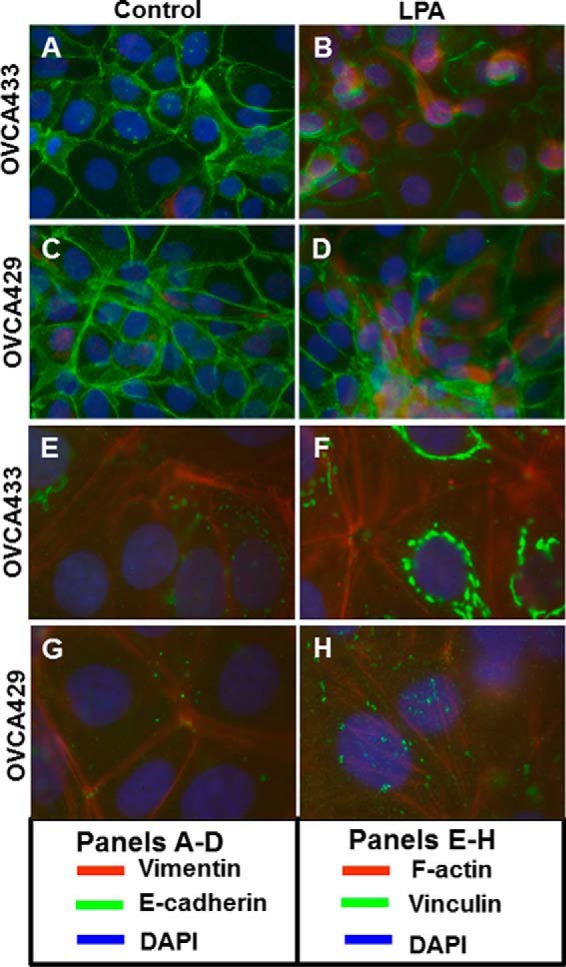
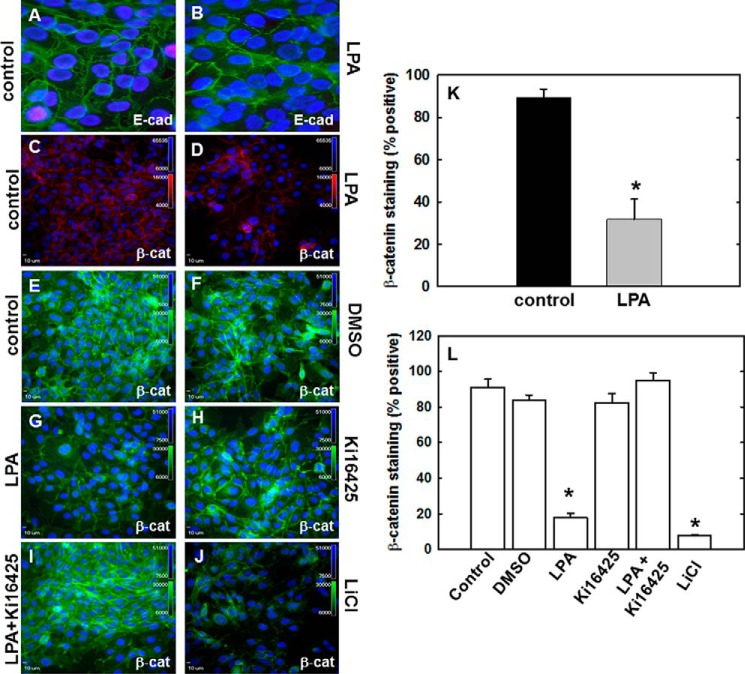
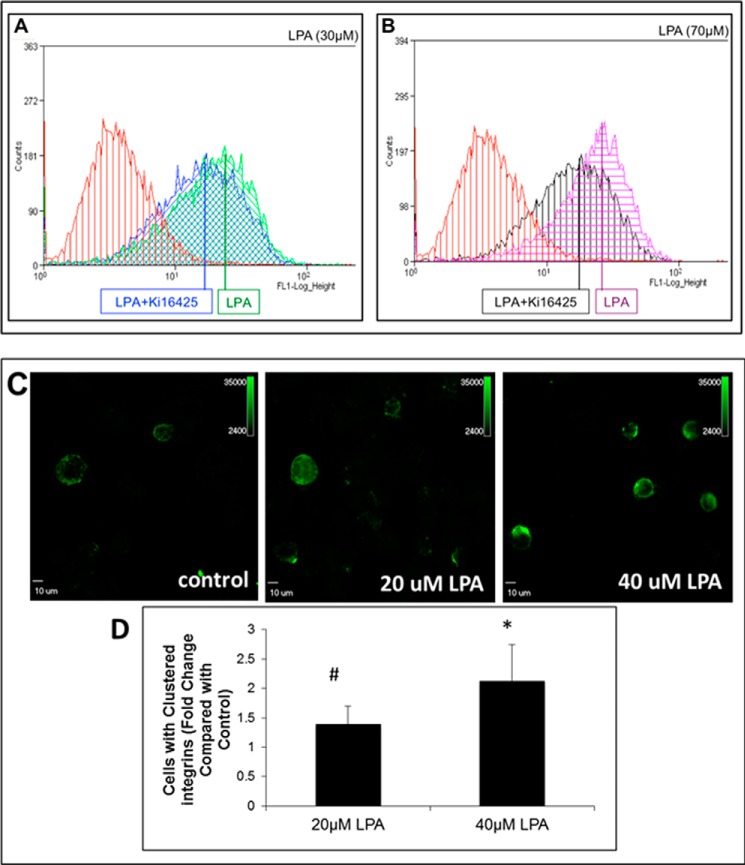
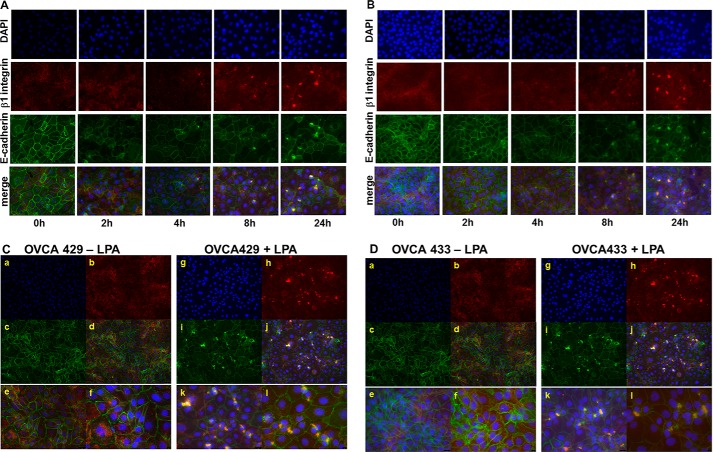
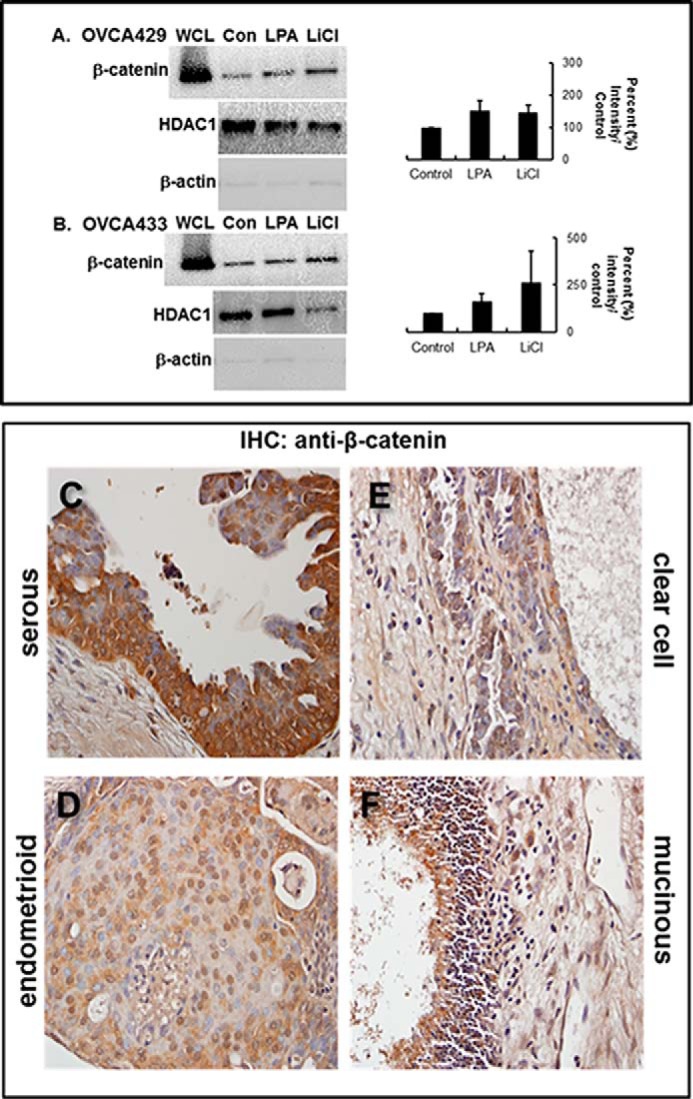
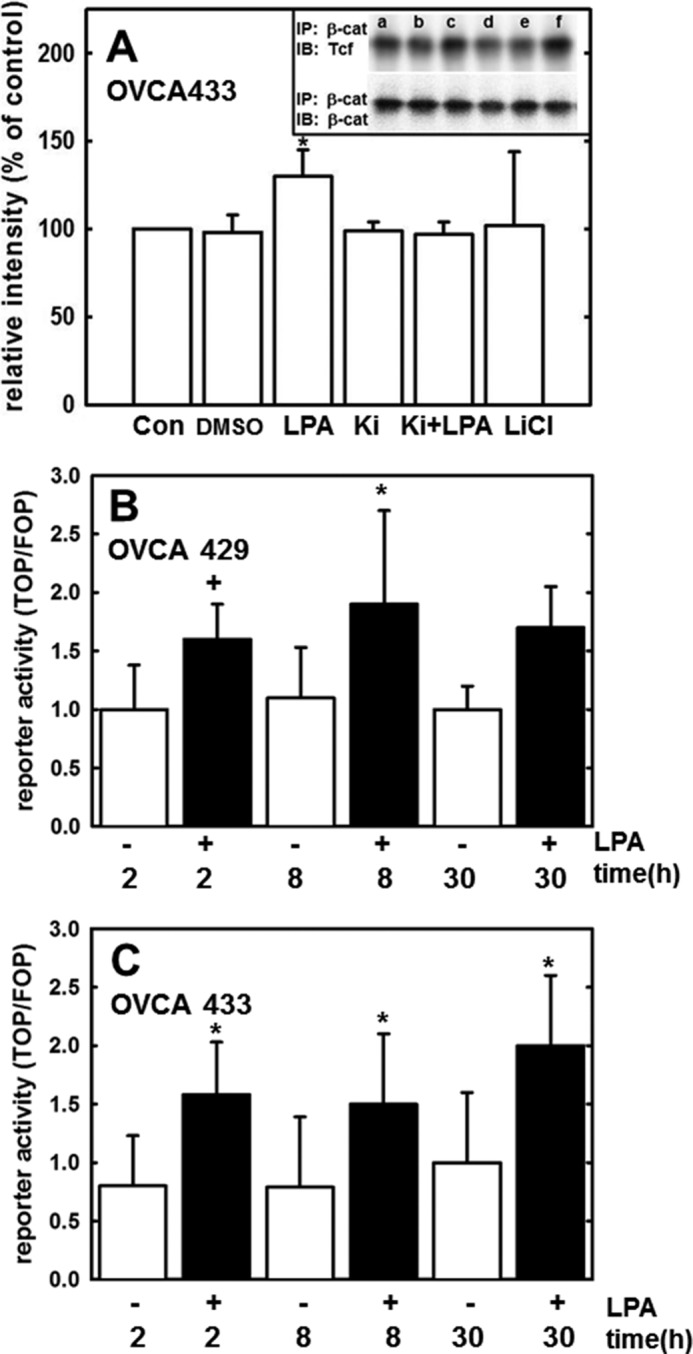
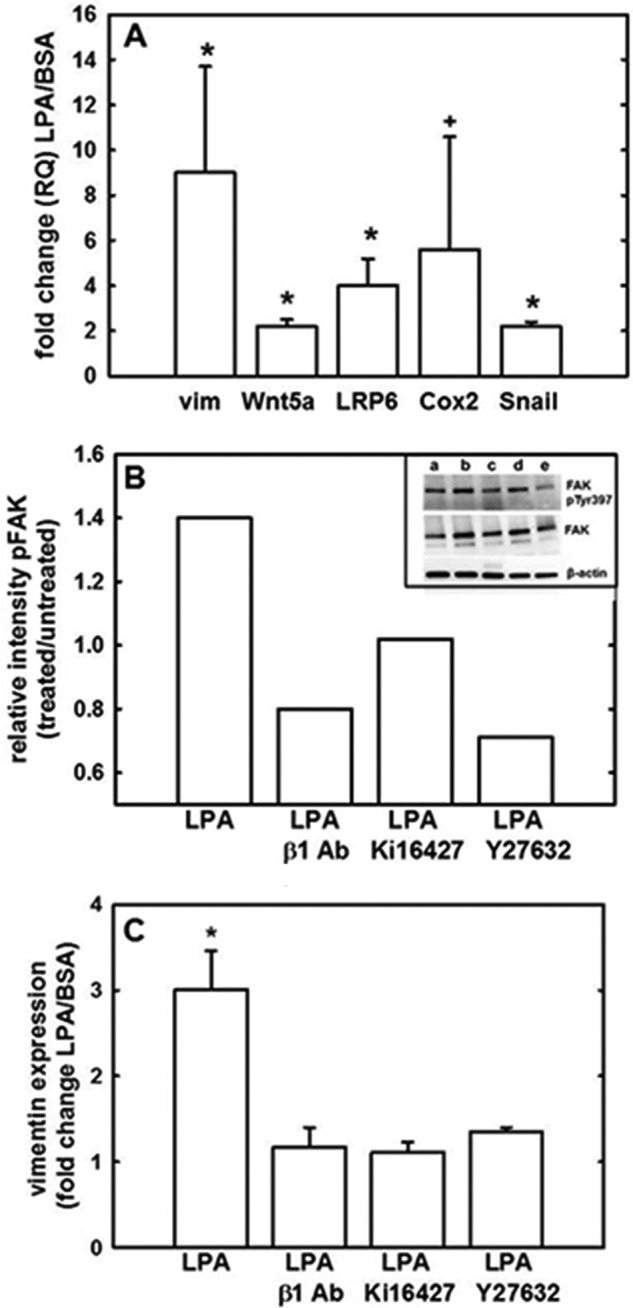
During tumor progression, epithelial ovarian cancer (EOC) cells undergo epithelial-to-mesenchymal transition (EMT), which influences metastatic success. Mutation-dependent activation of Wnt/β-catenin signaling has been implicated in gain of mesenchymal phenotype and loss of differentiation in several solid tumors; however, similar mutations are rare in most EOC histotypes. Nevertheless, evidence for activated Wnt/β-catenin signaling in EOC has been reported, and immunohistochemical analysis of human EOC tumors demonstrates nuclear staining in all histotypes. This study addresses the hypothesis that the bioactive lipid lysophosphatidic acid (LPA), prevalent in the EOC microenvironment, functions to regulate EMT in EOC. Our results demonstrate that LPA induces loss of junctional β-catenin, stimulates clustering of β1 integrins, and enhances the conformationally active population of surface β1 integrins. Furthermore, LPA treatment initiates nuclear translocation of β-catenin and transcriptional activation of Wnt/β-catenin target genes resulting in gain of mesenchymal marker expression. Together these data suggest that LPA initiates EMT in ovarian tumors through β1-integrin-dependent activation of Wnt/β-catenin signaling, providing a novel mechanism for mutation-independent activation of this pathway in EOC progression.
Keywords: LPA; Wnt signaling; integrin; lysophospholipid; ovarian cancer; β-catenin (β-catenin).
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Cannistra S. A. (2004) Cancer of the ovary. N. Engl. J. Med. 351, 2519–2529 - PubMed
-
- Seidman J. D., Horkayne-Szakaly I., Haiba M., Boice C. R., Kurman R. J., Ronnett B. M. (2004) The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int. J. Gynecol. Pathol. 23, 41–44 - PubMed
-
- American Cancer Society (2010) Cancer facts & figures. American Cancer Society, Atlanta, GA
-
- Surveillance Research Program, NCI (2014) SEER Stat Fact Sheets: Ovary cancer. National Cancer Institute, Bethesda
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
