

SNBRFinder: A Sequence-Based Hybrid Algorithm for Enhanced Prediction of Nucleic Acid-Binding Residues
- PMID: 26176857
- PMCID: PMC4503397
- DOI: 10.1371/journal.pone.0133260
SNBRFinder: A Sequence-Based Hybrid Algorithm for Enhanced Prediction of Nucleic Acid-Binding Residues
Abstract
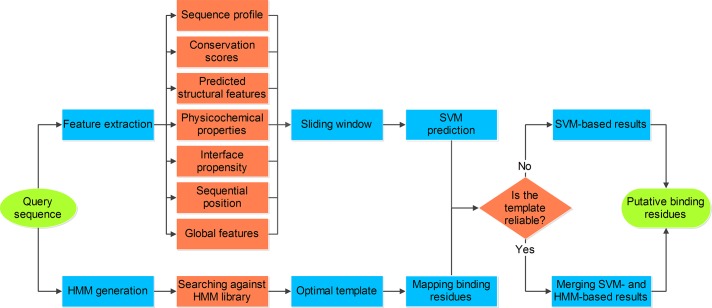
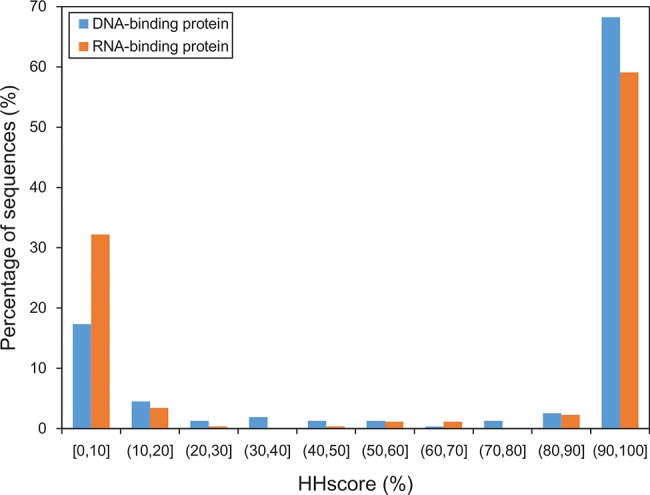
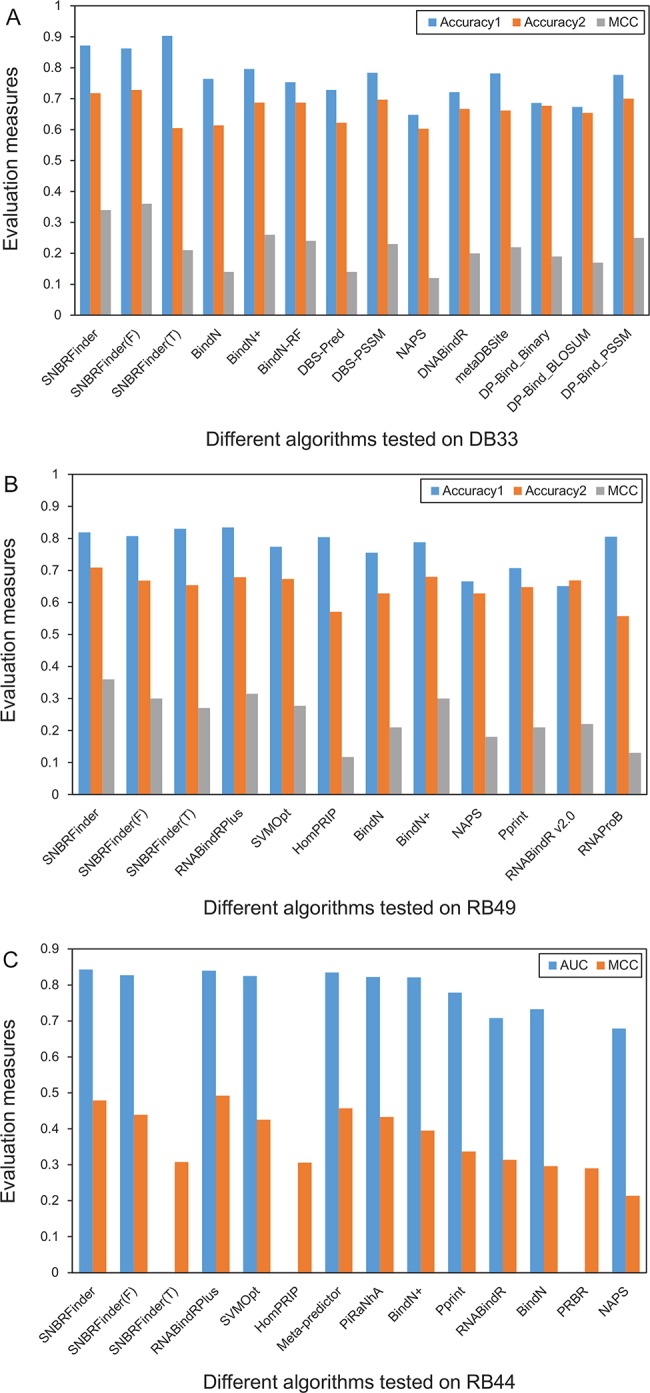
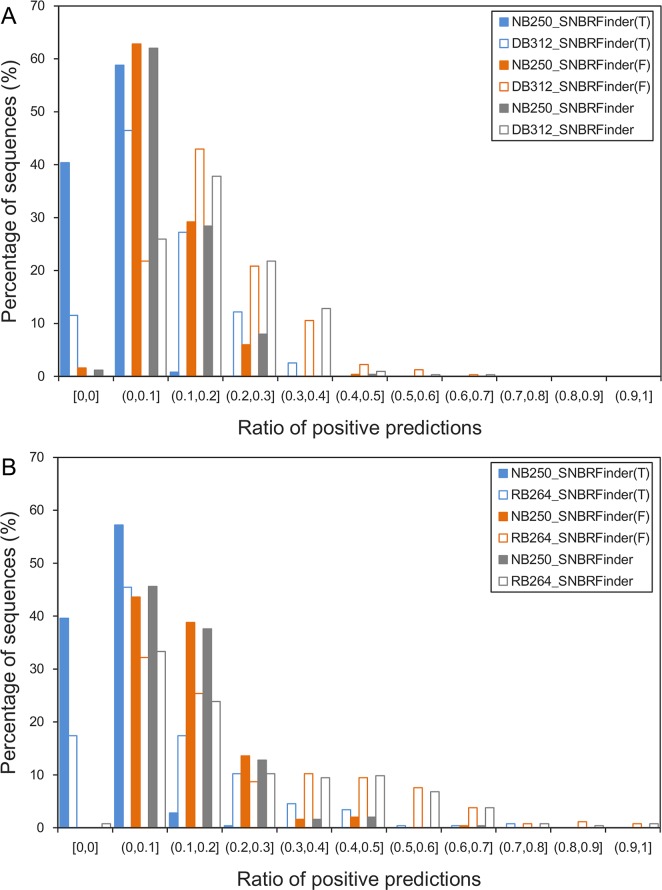
Protein-nucleic acid interactions are central to various fundamental biological processes. Automated methods capable of reliably identifying DNA- and RNA-binding residues in protein sequence are assuming ever-increasing importance. The majority of current algorithms rely on feature-based prediction, but their accuracy remains to be further improved. Here we propose a sequence-based hybrid algorithm SNBRFinder (Sequence-based Nucleic acid-Binding Residue Finder) by merging a feature predictor SNBRFinderF and a template predictor SNBRFinderT. SNBRFinderF was established using the support vector machine whose inputs include sequence profile and other complementary sequence descriptors, while SNBRFinderT was implemented with the sequence alignment algorithm based on profile hidden Markov models to capture the weakly homologous template of query sequence. Experimental results show that SNBRFinderF was clearly superior to the commonly used sequence profile-based predictor and SNBRFinderT can achieve comparable performance to the structure-based template methods. Leveraging the complementary relationship between these two predictors, SNBRFinder reasonably improved the performance of both DNA- and RNA-binding residue predictions. More importantly, the sequence-based hybrid prediction reached competitive performance relative to our previous structure-based counterpart. Our extensive and stringent comparisons show that SNBRFinder has obvious advantages over the existing sequence-based prediction algorithms. The value of our algorithm is highlighted by establishing an easy-to-use web server that is freely accessible at http://ibi.hzau.edu.cn/SNBRFinder.
Conflict of interest statement
Figures

Similar articles
-
RBRDetector: improved prediction of binding residues on RNA-binding protein structures using complementary feature- and template-based strategies.Proteins. 2014 Oct;82(10):2455-71. doi: 10.1002/prot.24610. Epub 2014 Jun 9. Proteins. 2014. PMID: 24854765
-
DNABind: a hybrid algorithm for structure-based prediction of DNA-binding residues by combining machine learning- and template-based approaches.Proteins. 2013 Nov;81(11):1885-99. doi: 10.1002/prot.24330. Epub 2013 Aug 16. Proteins. 2013. PMID: 23737141
-
NdPASA: a novel pairwise protein sequence alignment algorithm that incorporates neighbor-dependent amino acid propensities.Proteins. 2005 Feb 15;58(3):628-37. doi: 10.1002/prot.20359. Proteins. 2005. PMID: 15616964
-
A comprehensive comparative review of sequence-based predictors of DNA- and RNA-binding residues.Brief Bioinform. 2016 Jan;17(1):88-105. doi: 10.1093/bib/bbv023. Epub 2015 May 1. Brief Bioinform. 2016. PMID: 25935161 Review.
-
Structural protein descriptors in 1-dimension and their sequence-based predictions.Curr Protein Pept Sci. 2011 Sep;12(6):470-89. doi: 10.2174/138920311796957711. Curr Protein Pept Sci. 2011. PMID: 21787299 Review.
Cited by
-
Twenty years of advances in prediction of nucleic acid-binding residues in protein sequences.Brief Bioinform. 2024 Nov 22;26(1):bbaf016. doi: 10.1093/bib/bbaf016. Brief Bioinform. 2024. PMID: 39833102 Free PMC article. Review.
-
Predictive modeling of moonlighting DNA-binding proteins.NAR Genom Bioinform. 2022 Dec 2;4(4):lqac091. doi: 10.1093/nargab/lqac091. eCollection 2022 Dec. NAR Genom Bioinform. 2022. PMID: 36474806 Free PMC article.
-
Precise prediction of phase-separation key residues by machine learning.Nat Commun. 2024 Mar 26;15(1):2662. doi: 10.1038/s41467-024-46901-9. Nat Commun. 2024. PMID: 38531854 Free PMC article.
-
A boosting approach for prediction of protein-RNA binding residues.BMC Bioinformatics. 2017 Dec 1;18(Suppl 13):465. doi: 10.1186/s12859-017-1879-2. BMC Bioinformatics. 2017. PMID: 29219069 Free PMC article.
-
Multi-Agent Systems for Resource Allocation and Scheduling in a Smart Grid.Sensors (Basel). 2022 Oct 22;22(21):8099. doi: 10.3390/s22218099. Sensors (Basel). 2022. PMID: 36365795 Free PMC article. Review.
References
-
- Chen Y, Varani G. Protein families and RNA recognition. FEBS J. 2005;272: 2088–97. - PubMed
-
- Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nature genetics. 2000;25: 192–4. - PubMed
-
- Ahmad S, Gromiha MM, Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20: 477–86. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
