

Distance-Based Configurational Entropy of Proteins from Molecular Dynamics Simulations
- PMID: 26177039
- PMCID: PMC4503633
- DOI: 10.1371/journal.pone.0132356
Distance-Based Configurational Entropy of Proteins from Molecular Dynamics Simulations
Abstract
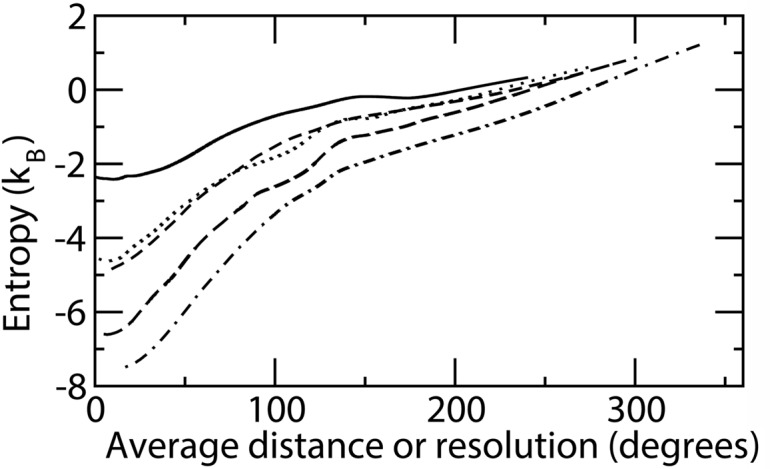
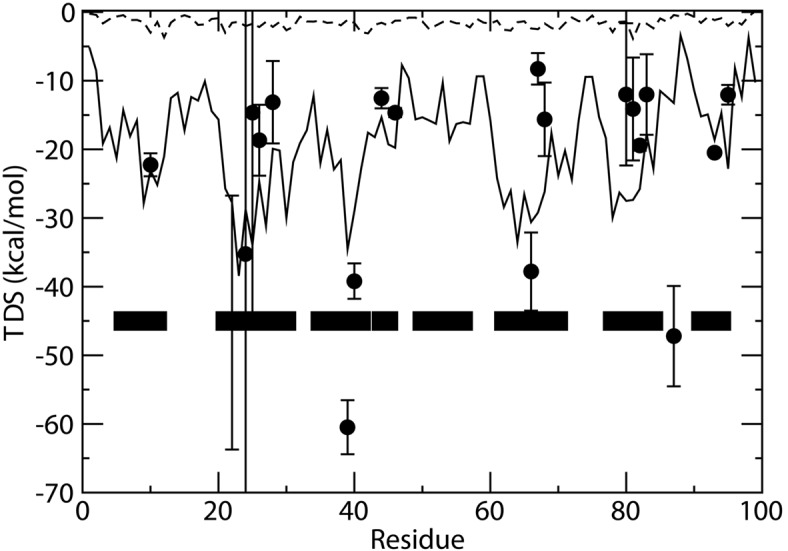
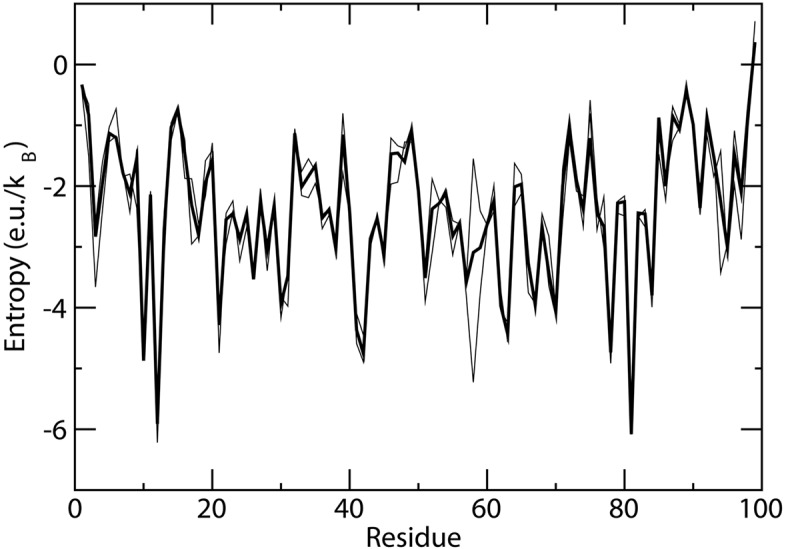
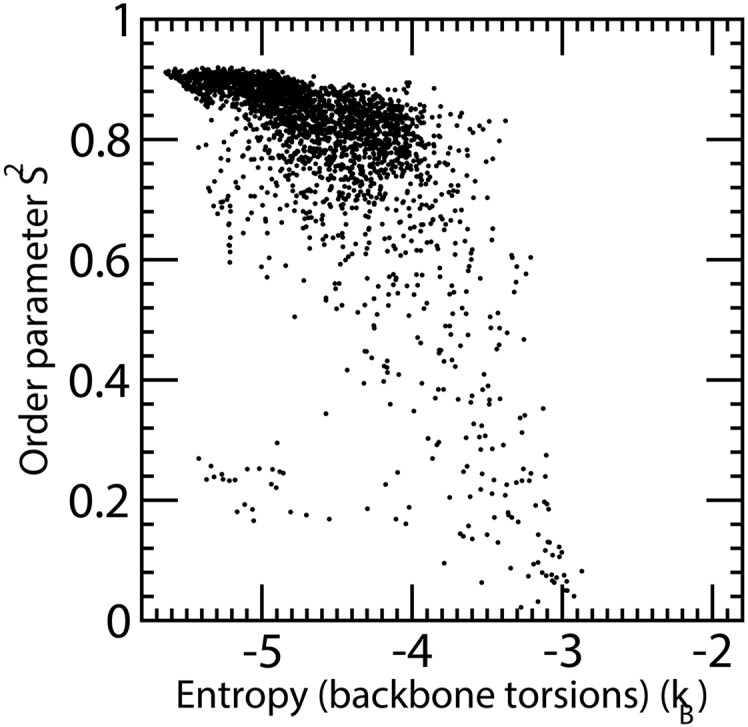
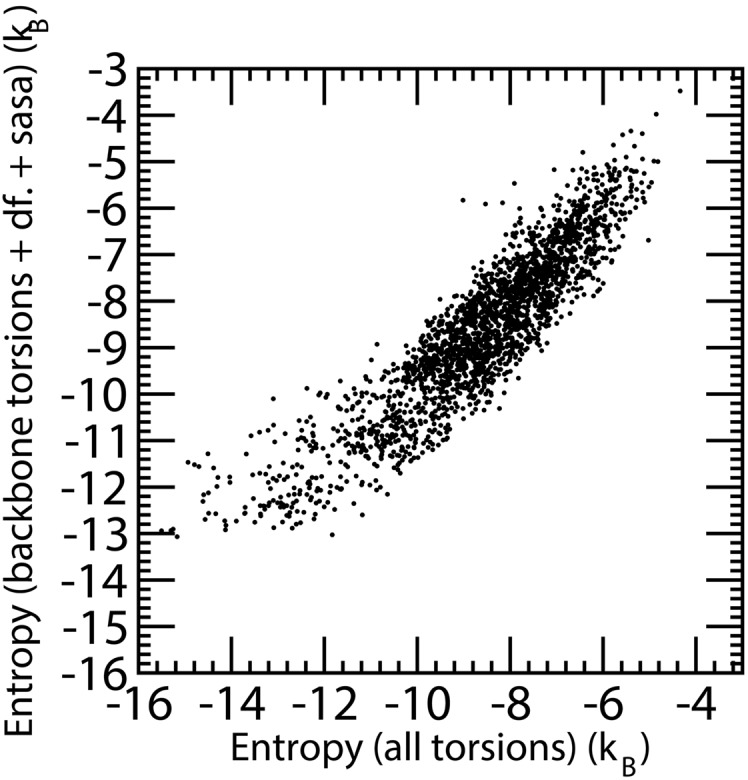
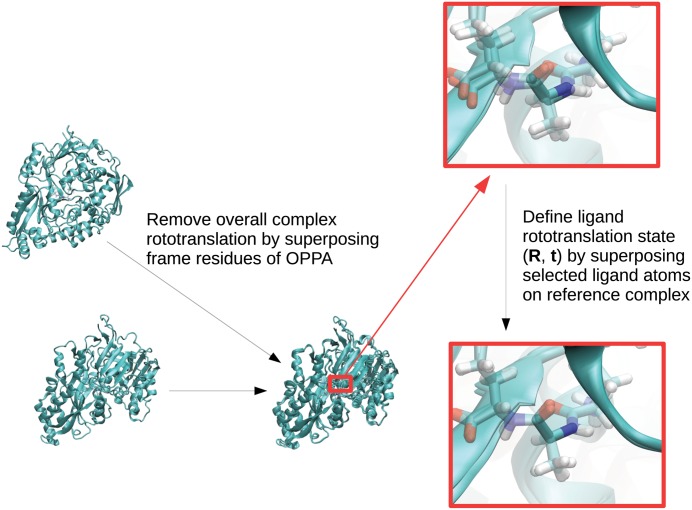
Estimation of configurational entropy from molecular dynamics trajectories is a difficult task which is often performed using quasi-harmonic or histogram analysis. An entirely different approach, proposed recently, estimates local density distribution around each conformational sample by measuring the distance from its nearest neighbors. In this work we show this theoretically well grounded the method can be easily applied to estimate the entropy from conformational sampling. We consider a set of systems that are representative of important biomolecular processes. In particular: reference entropies for amino acids in unfolded proteins are obtained from a database of residues not participating in secondary structure elements;the conformational entropy of folding of β2-microglobulin is computed from molecular dynamics simulations using reference entropies for the unfolded state;backbone conformational entropy is computed from molecular dynamics simulations of four different states of the EPAC protein and compared with order parameters (often used as a measure of entropy);the conformational and rototranslational entropy of binding is computed from simulations of 20 tripeptides bound to the peptide binding protein OppA and of β2-microglobulin bound to a citrate coated gold surface. This work shows the potential of the method in the most representative biological processes involving proteins, and provides a valuable alternative, principally in the shown cases, where other approaches are problematic.
Conflict of interest statement
Figures
Similar articles
-
The Dynameomics Entropy Dictionary: A Large-Scale Assessment of Conformational Entropy across Protein Fold Space.J Phys Chem B. 2017 Apr 27;121(16):3933-3945. doi: 10.1021/acs.jpcb.7b00577. Epub 2017 Apr 19. J Phys Chem B. 2017. PMID: 28375008
-
Dissecting Protein Configurational Entropy into Conformational and Vibrational Contributions.J Phys Chem B. 2015 Oct 1;119(39):12623-31. doi: 10.1021/acs.jpcb.5b07060. Epub 2015 Sep 16. J Phys Chem B. 2015. PMID: 26348368
-
Accurate Estimation of the Entropy of Rotation-Translation Probability Distributions.J Chem Theory Comput. 2016 Jan 12;12(1):1-8. doi: 10.1021/acs.jctc.5b00731. Epub 2015 Dec 10. J Chem Theory Comput. 2016. PMID: 26605696
-
Markov state models provide insights into dynamic modulation of protein function.Acc Chem Res. 2015 Feb 17;48(2):414-22. doi: 10.1021/ar5002999. Epub 2015 Jan 3. Acc Chem Res. 2015. PMID: 25625937 Free PMC article. Review.
-
Side-chain conformational entropy in protein folding.Protein Sci. 1995 Nov;4(11):2247-51. doi: 10.1002/pro.5560041101. Protein Sci. 1995. PMID: 8563620 Free PMC article. Review.
Cited by
-
Free Energy, Enthalpy and Entropy from Implicit Solvent End-Point Simulations.Front Mol Biosci. 2018 Feb 8;5:11. doi: 10.3389/fmolb.2018.00011. eCollection 2018. Front Mol Biosci. 2018. PMID: 29473043 Free PMC article.
-
Recent Advances in EPAC-Targeted Therapies: A Biophysical Perspective.Cells. 2019 Nov 19;8(11):1462. doi: 10.3390/cells8111462. Cells. 2019. PMID: 31752286 Free PMC article. Review.
-
A Rationalization of the Effect That TMAO, Glycine, and Betaine Exert on the Collapse of Elastin-like Polypeptides.Life (Basel). 2022 Jan 18;12(2):140. doi: 10.3390/life12020140. Life (Basel). 2022. PMID: 35207427 Free PMC article.
-
Dynamics and Thermodynamics of Transthyretin Association from Molecular Dynamics Simulations.Biomed Res Int. 2018 Jun 5;2018:7480749. doi: 10.1155/2018/7480749. eCollection 2018. Biomed Res Int. 2018. PMID: 29967786 Free PMC article.
-
On the Stabilizing Effect of Aspartate and Glutamate and Its Counteraction by Common Denaturants.Int J Mol Sci. 2024 Aug 29;25(17):9360. doi: 10.3390/ijms25179360. Int J Mol Sci. 2024. PMID: 39273310 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
