

D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2
- PMID: 26178471
- PMCID: PMC4515030
- DOI: 10.1038/ncomms8768
D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2
Abstract
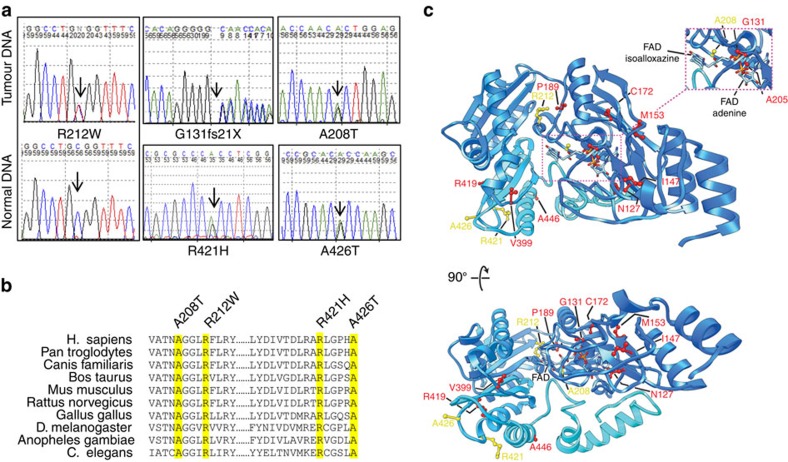
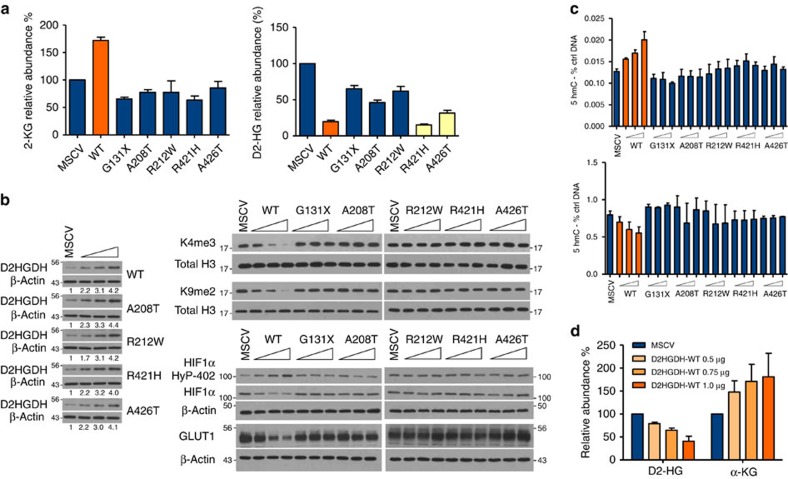
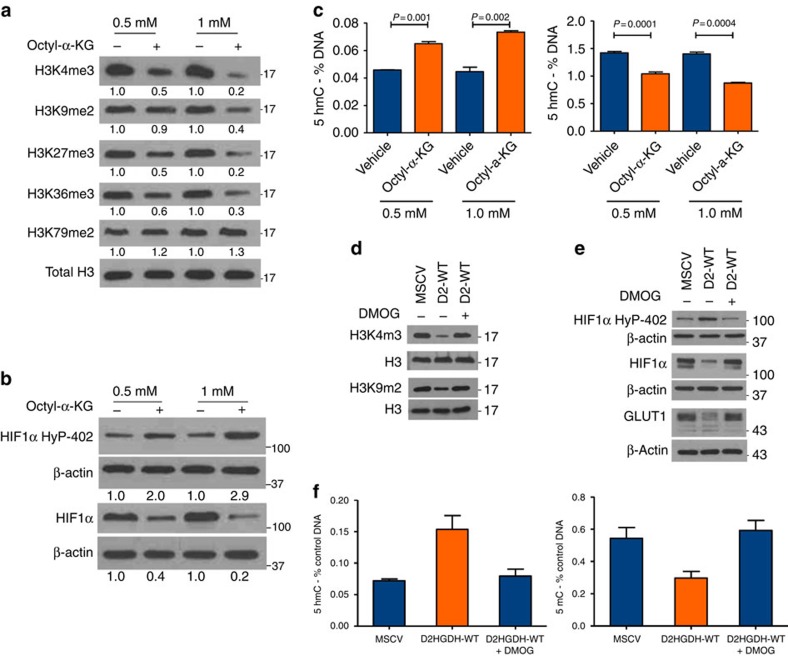
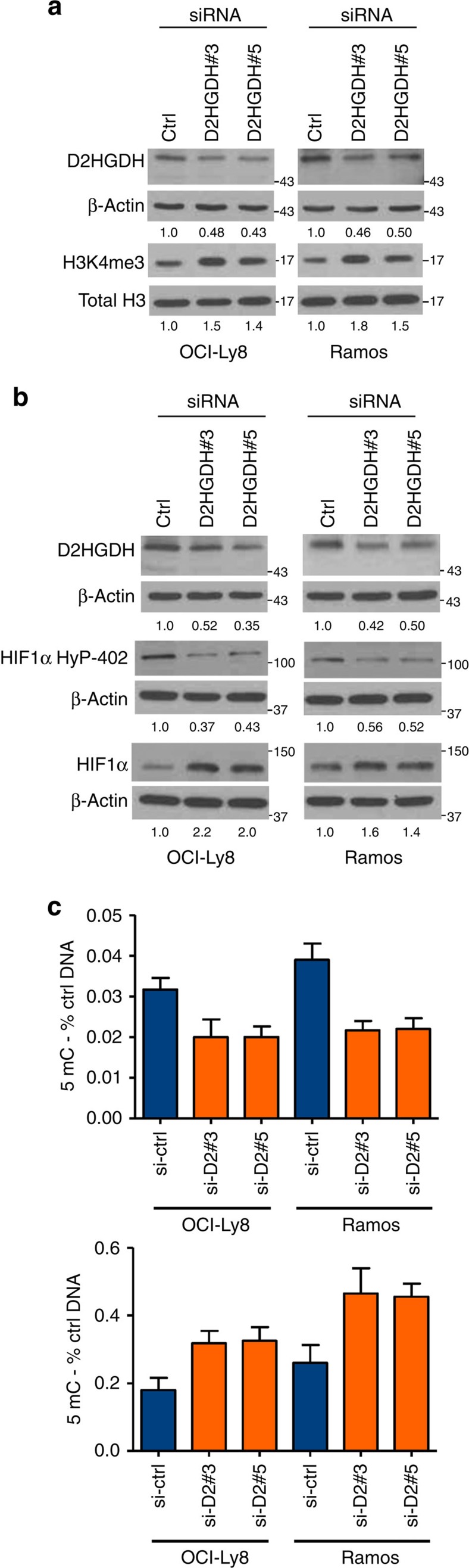
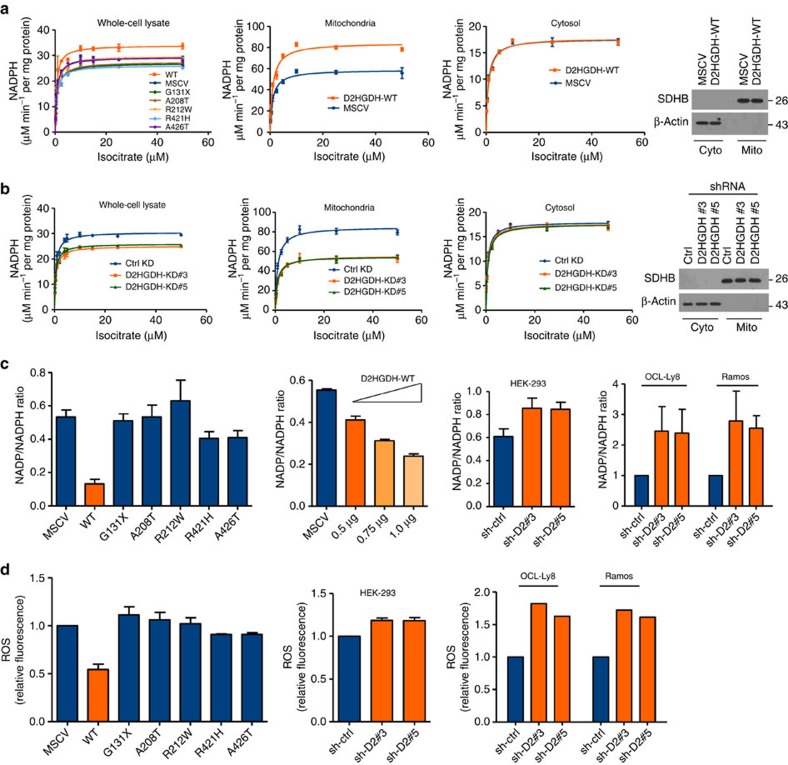
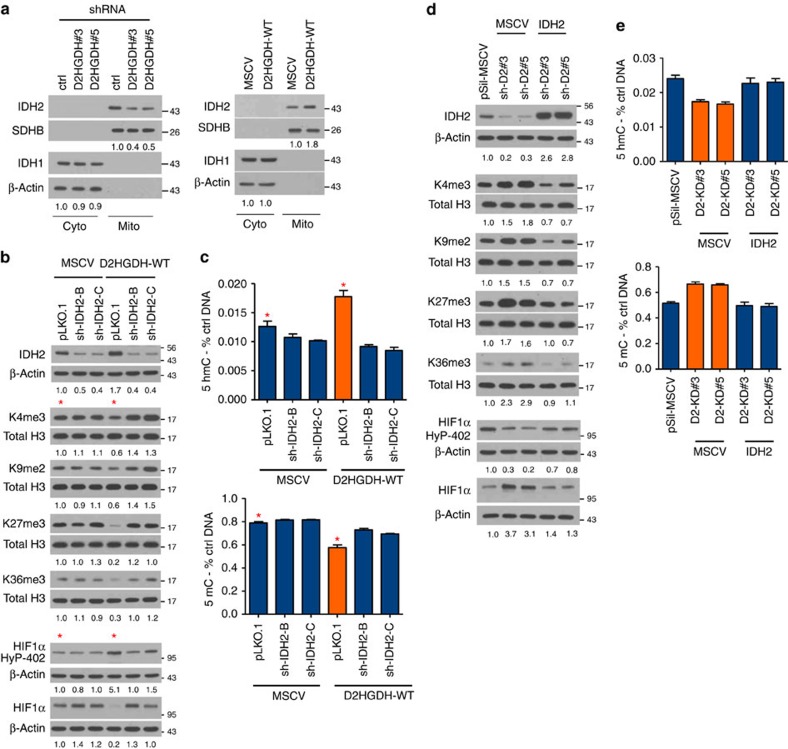
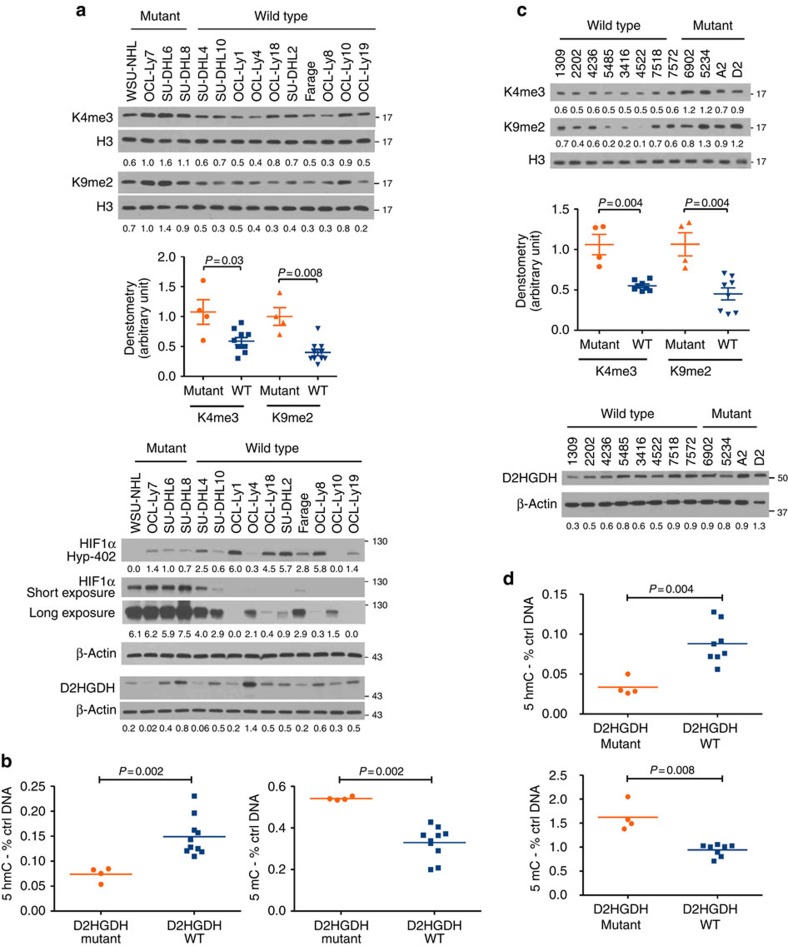
Isocitrate dehydrogenases (IDH) convert isocitrate to alpha-ketoglutarate (α-KG). In cancer, mutant IDH1/2 reduces α-KG to D2-hydroxyglutarate (D2-HG) disrupting α-KG-dependent dioxygenases. However, the physiological relevance of controlling the interconversion of D2-HG into α-KG, mediated by D2-hydroxyglutarate dehydrogenase (D2HGDH), remains obscure. Here we show that wild-type D2HGDH elevates α-KG levels, influencing histone and DNA methylation, and HIF1α hydroxylation. Conversely, the D2HGDH mutants that we find in diffuse large B-cell lymphoma are enzymatically inert. D2-HG is a low-abundance metabolite, but we show that it can meaningfully elevate α-KG levels by positively modulating mitochondrial IDH activity and inducing IDH2 expression. Accordingly, genetic depletion of IDH2 abrogates D2HGDH effects, whereas ectopic IDH2 rescues D2HGDH-deficient cells. Our data link D2HGDH to cancer and describe an additional role for the enzyme: the regulation of IDH2 activity and α-KG-mediated epigenetic remodelling. These data further expose the intricacies of mitochondrial metabolism and inform on the pathogenesis of D2HGDH-deficient diseases.
Figures
References
-
- Loenarz C. & Schofield C. J. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 4, 152–156 (2008) . - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
