

Myelin-associated glycoprotein gene mutation causes Pelizaeus-Merzbacher disease-like disorder
- PMID: 26179919
- PMCID: PMC4643626
- DOI: 10.1093/brain/awv204
Myelin-associated glycoprotein gene mutation causes Pelizaeus-Merzbacher disease-like disorder
Abstract
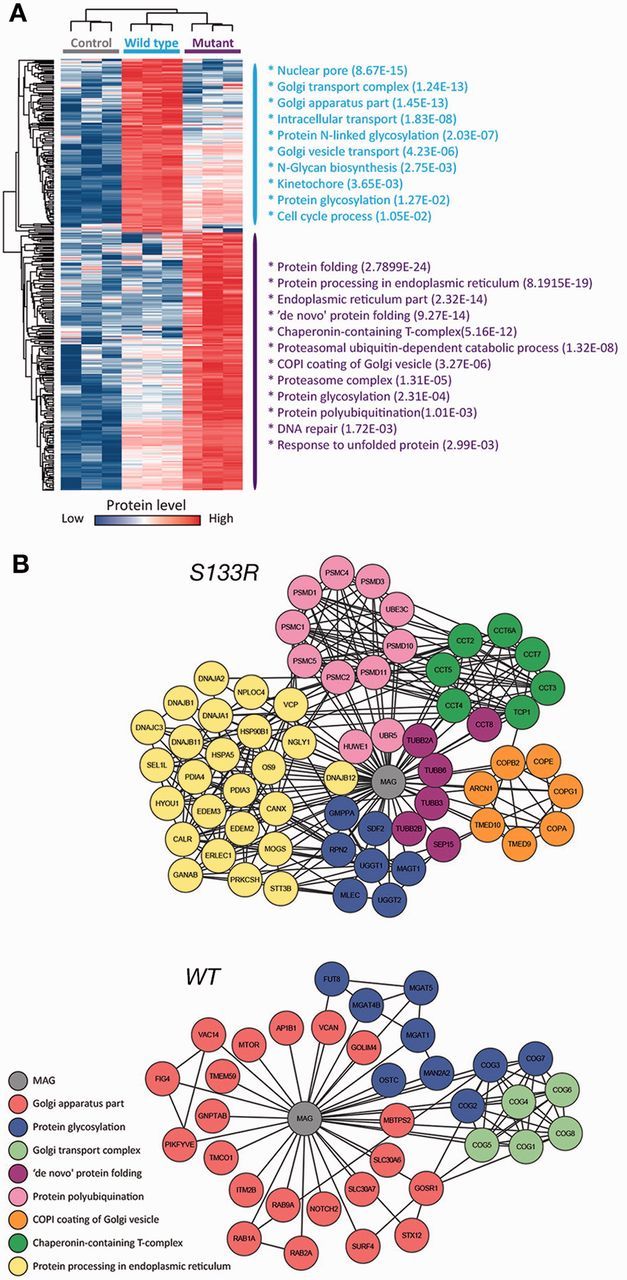
Pelizaeus-Merzbacher disease is an X-linked hypomyelinating leukodystrophy caused by mutations or rearrangements in PLP1. It presents in infancy with nystagmus, jerky head movements, hypotonia and developmental delay evolving into spastic tetraplegia with optic atrophy and variable movement disorders. A clinically similar phenotype caused by recessive mutations in GJC2 is known as Pelizaeus-Merzbacher-like disease. Both genes encode proteins associated with myelin. We describe three siblings of a consanguineous family manifesting the typical infantile-onset Pelizaeus-Merzbacher disease-like phenotype slowly evolving into a form of complicated hereditary spastic paraplegia with mental retardation, dysarthria, optic atrophy and peripheral neuropathy in adulthood. Magnetic resonance imaging and spectroscopy were consistent with a demyelinating leukodystrophy. Using genetic linkage and exome sequencing, we identified a homozygous missense c.399C>G; p.S133R mutation in MAG. This gene, previously associated with hereditary spastic paraplegia, encodes myelin-associated glycoprotein, which is involved in myelin maintenance and glia-axon interaction. This mutation is predicted to destabilize the protein and affect its tertiary structure. Examination of the sural nerve biopsy sample obtained in childhood in the oldest sibling revealed complete absence of myelin-associated glycoprotein accompanied by ill-formed onion-bulb structures and a relatively thin myelin sheath of the affected axons. Immunofluorescence, cell surface labelling, biochemical analysis and mass spectrometry-based proteomics studies in a variety of cell types demonstrated a devastating effect of the mutation on post-translational processing, steady state expression and subcellular localization of myelin-associated glycoprotein. In contrast to the wild-type protein, the p.S133R mutant was retained in the endoplasmic reticulum and was subjected to endoplasmic reticulum-associated protein degradation by the proteasome. Our findings identify involvement of myelin-associated glycoprotein in this family with a disorder affecting the central and peripheral nervous system, and suggest that loss of the protein function is responsible for the unique clinical phenotype.
Keywords: MAG; Pelizaeus-Merzbacher-like disease; hereditary spastic paraplegia.
© The Author (2015). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures







Similar articles
-
Inherited white matter disorders: Hypomyelination (myelin disorders).Handb Clin Neurol. 2024;204:197-223. doi: 10.1016/B978-0-323-99209-1.00014-4. Handb Clin Neurol. 2024. PMID: 39322379 Review.
-
Pelizaeus-Merzbacher disease and spastic paraplegia type 2: two faces of myelin loss from mutations in the same gene.J Child Neurol. 2003 Sep;18(9):616-24. doi: 10.1177/08830738030180090801. J Child Neurol. 2003. PMID: 14572140 Review.
-
The spectrum of PLP1 gene mutations in patients with the classical form of the Pelizaeus-Merzbacher disease.Med Wieku Rozwoj. 2013 Oct-Dec;17(4):293-300. Med Wieku Rozwoj. 2013. PMID: 24519770
-
Novel mutations in the GJC2 gene associated with Pelizaeus-Merzbacher-like disease.Mol Biol Rep. 2019 Aug;46(4):4507-4516. doi: 10.1007/s11033-019-04906-4. Epub 2019 Jul 3. Mol Biol Rep. 2019. PMID: 31270756
-
Three new PLP1 splicing mutations demonstrate pathogenic and phenotypic diversity of Pelizaeus-Merzbacher disease.J Child Neurol. 2014 Jul;29(7):924-31. doi: 10.1177/0883073813492387. Epub 2013 Jun 14. J Child Neurol. 2014. PMID: 23771846
Cited by
-
Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia.Brain Res. 2016 Oct 1;1648(Pt B):594-602. doi: 10.1016/j.brainres.2016.03.046. Epub 2016 Apr 4. Brain Res. 2016. PMID: 27055915 Free PMC article. Review.
-
Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: implications in frontotemporal lobar degeneration.Aging (Albany NY). 2017 Mar 9;9(3):823-851. doi: 10.18632/aging.101195. Aging (Albany NY). 2017. PMID: 28283675 Free PMC article.
-
Discovery, classification, evolution and diversity of Siglecs.Mol Aspects Med. 2023 Apr;90:101117. doi: 10.1016/j.mam.2022.101117. Epub 2022 Aug 18. Mol Aspects Med. 2023. PMID: 35989204 Free PMC article. Review.
-
Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders.Front Mol Neurosci. 2017 Jan 4;9:162. doi: 10.3389/fnmol.2016.00162. eCollection 2016. Front Mol Neurosci. 2017. PMID: 28101003 Free PMC article. Review.
-
Novel Mutations in NPC1 are Associated with Pelizaeus-Merzbacher-Like Disease: A Case Report.Int J Gen Med. 2021 Mar 9;14:797-803. doi: 10.2147/IJGM.S293675. eCollection 2021. Int J Gen Med. 2021. PMID: 33727856 Free PMC article.
References
-
- Altier C, Garcia-Caballero A, Simms B, You H, Chen L, Walcher J, et al. The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat Neurosci 2011; 14: 173–80. - PubMed
-
- Burger D, Simon M, Perruisseau G, Steck AJ. The epitope(s) recognized by HNK-1 antibody and IgM paraprotein in neuropathy is present on several N-linked oligosaccharide structure on human P0 and myelin-associated glycoprotein. J Neurochem 1990; 54: 1569–75. - PubMed
-
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 2011; 10: 1794–805. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
