

Zeta Inhibitory Peptide Disrupts Electrostatic Interactions That Maintain Atypical Protein Kinase C in Its Active Conformation on the Scaffold p62
- PMID: 26187466
- PMCID: PMC4571940
- DOI: 10.1074/jbc.M115.676221
Zeta Inhibitory Peptide Disrupts Electrostatic Interactions That Maintain Atypical Protein Kinase C in Its Active Conformation on the Scaffold p62
Abstract
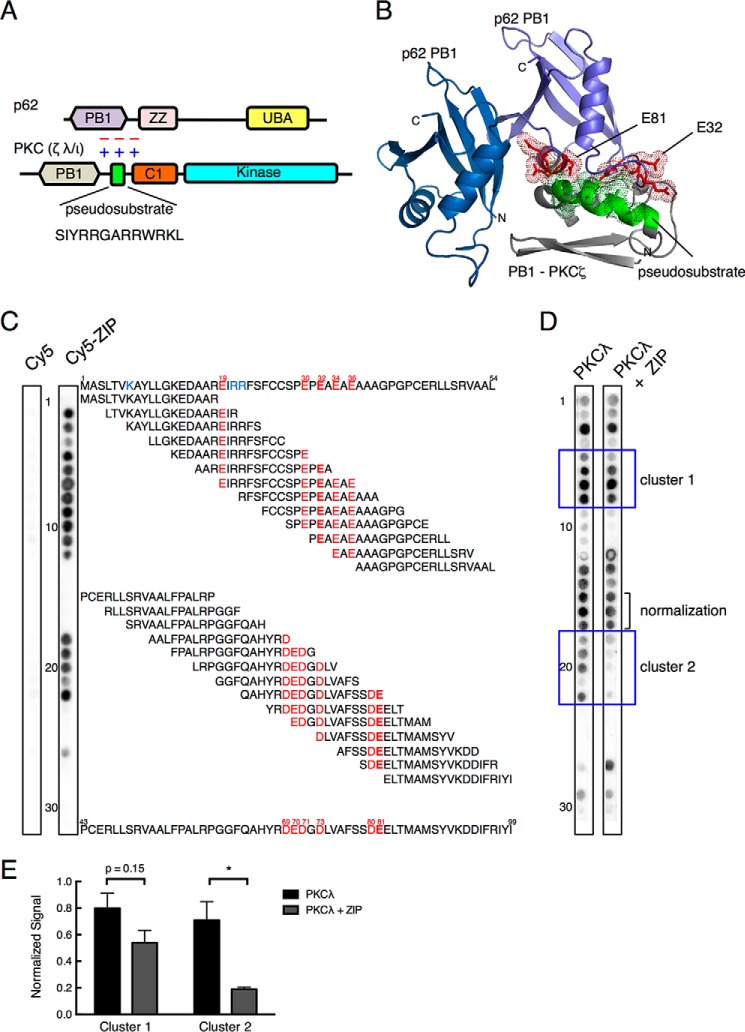
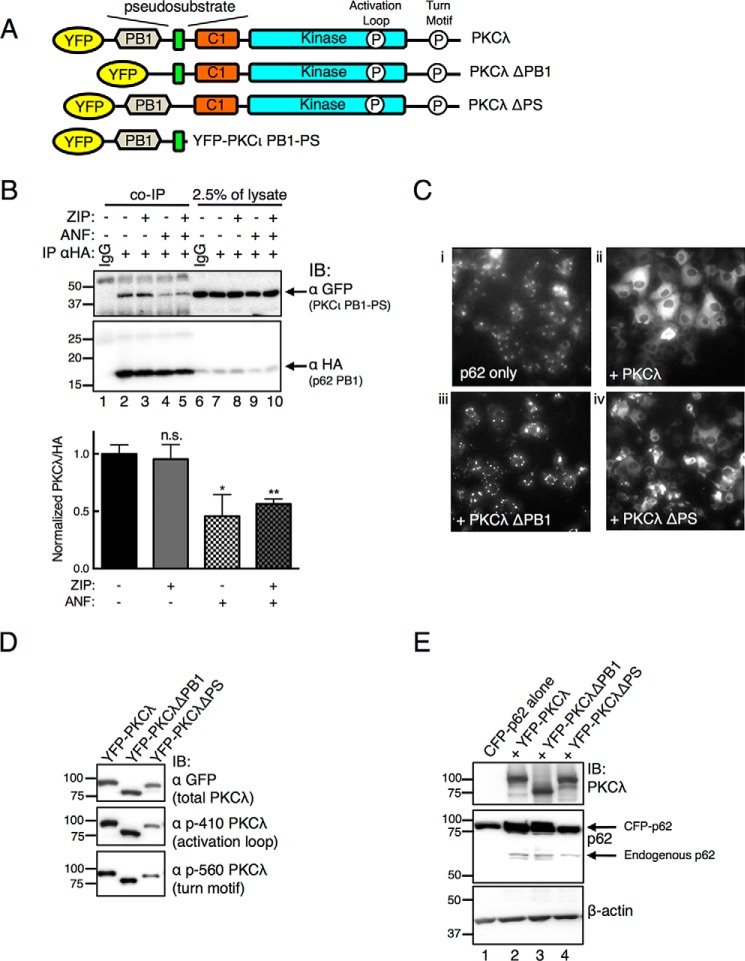
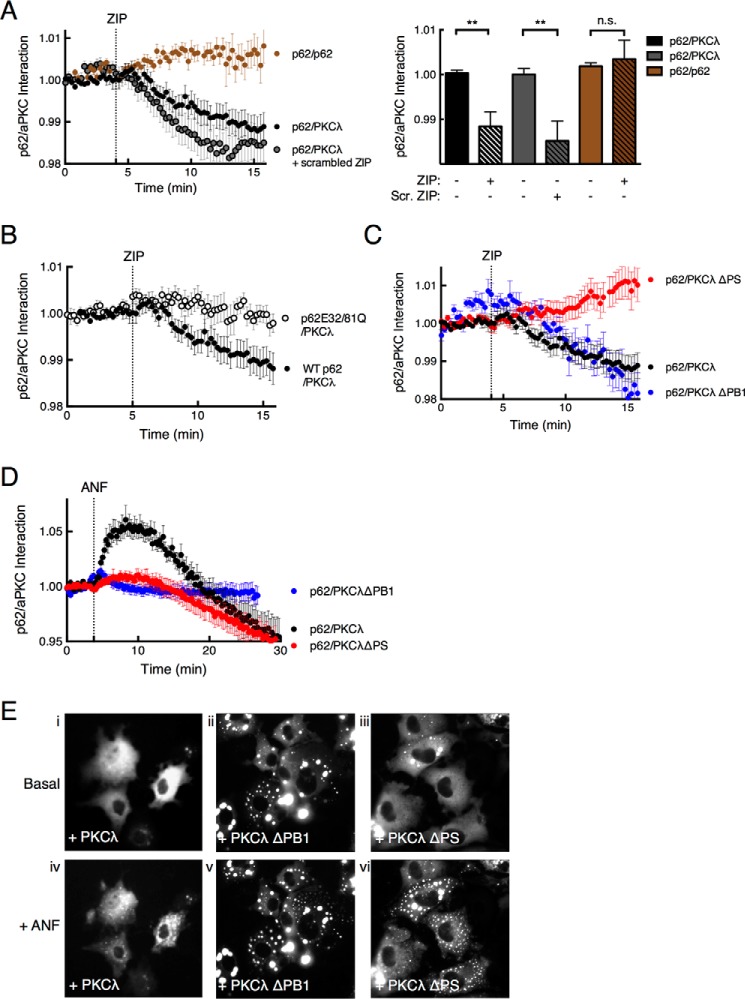
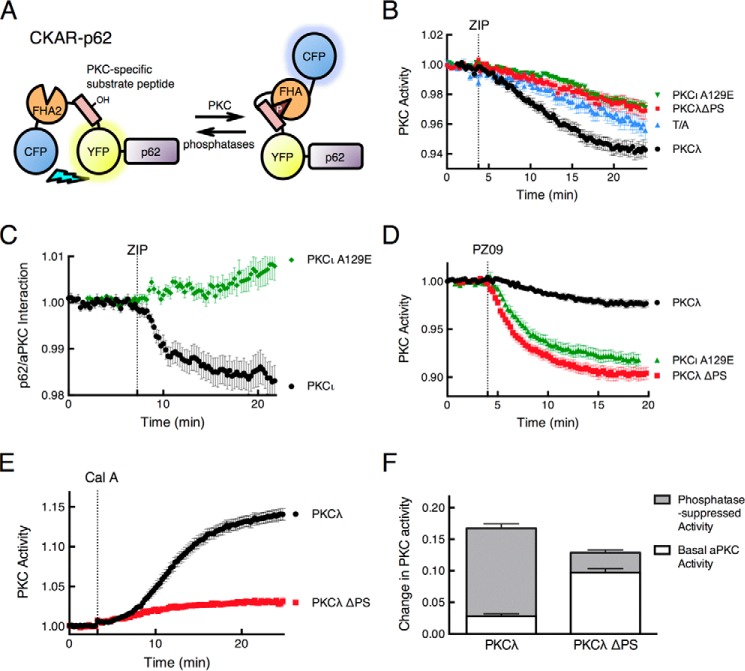
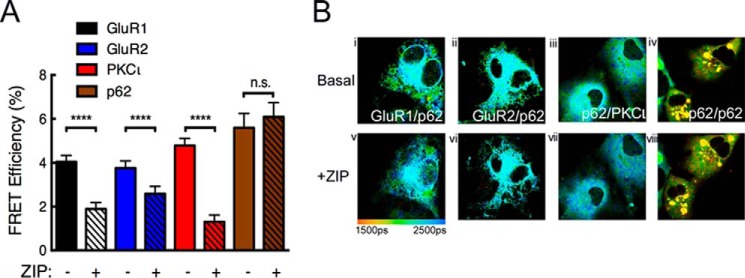
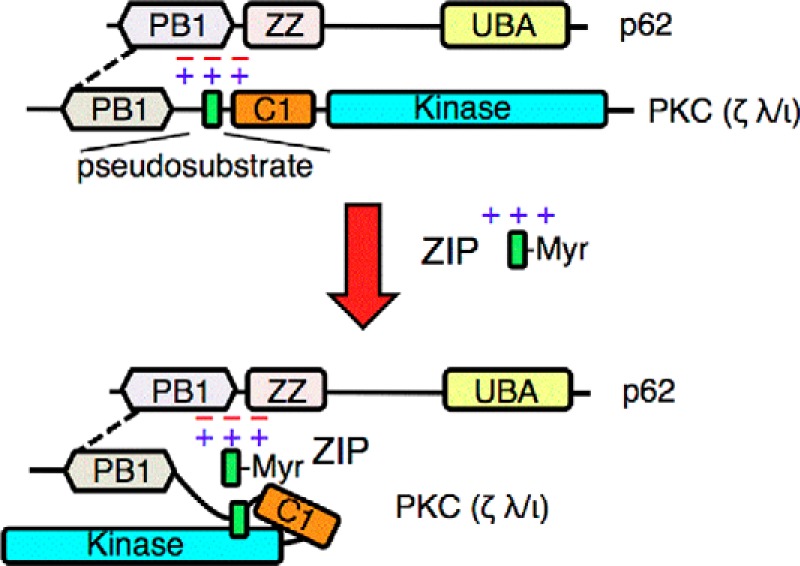
Atypical protein kinase C (aPKC) enzymes signal on protein scaffolds, yet how they are maintained in an active conformation on scaffolds is unclear. A myristoylated peptide based on the autoinhibitory pseudosubstrate fragment of the atypical PKCζ, zeta inhibitory peptide (ZIP), has been extensively used to inhibit aPKC activity; however, we have previously shown that ZIP does not inhibit the catalytic activity of aPKC isozymes in cells (Wu-Zhang, A. X., Schramm, C. L., Nabavi, S., Malinow, R., and Newton, A. C. (2012) J. Biol. Chem. 287, 12879-12885). Here we sought to identify a bona fide target of ZIP and, in so doing, unveiled a novel mechanism by which aPKCs are maintained in an active conformation on a protein scaffold. Specifically, we used protein-protein interaction network analysis, structural modeling, and protein-protein docking to predict that ZIP binds an acidic surface on the Phox and Bem1 (PB1) domain of p62, an interaction validated by peptide array analysis. Using a genetically encoded reporter for PKC activity fused to the p62 scaffold, we show that ZIP inhibits the activity of wild-type aPKC, but not a construct lacking the pseudosubstrate. These data support a model in which the pseudosubstrate of aPKCs is tethered to the acidic surface on p62, locking aPKC in an open, signaling-competent conformation. ZIP competes for binding to the acidic surface, resulting in displacement of the pseudosubstrate of aPKC and re-engagement in the substrate-binding cavity. This study not only identifies a cellular target for ZIP, but also unveils a novel mechanism by which scaffolded aPKC is maintained in an active conformation.
Keywords: atypical protein kinase C (aPKC); enzyme mechanism; p62 (sequestosome 1(SQSTM1)); protein-protein interaction; scaffold protein; serine/threonine protein kinase.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Protein Scaffolds Control Localized Protein Kinase Cζ Activity.J Biol Chem. 2016 Jun 24;291(26):13809-22. doi: 10.1074/jbc.M116.729483. Epub 2016 May 3. J Biol Chem. 2016. PMID: 27143478 Free PMC article.
-
Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5.J Biol Chem. 2004 Jul 23;279(30):31883-90. doi: 10.1074/jbc.M403092200. Epub 2004 May 13. J Biol Chem. 2004. PMID: 15143057
-
Protein Kinase C (PKC)ζ Pseudosubstrate Inhibitor Peptide Promiscuously Binds PKC Family Isoforms and Disrupts Conventional PKC Targeting and Translocation.Mol Pharmacol. 2015 Oct;88(4):728-35. doi: 10.1124/mol.115.099457. Epub 2015 Jul 21. Mol Pharmacol. 2015. PMID: 26199377 Free PMC article.
-
Targeting the oncogenic protein kinase Ciota signalling pathway for the treatment of cancer.Biochem Soc Trans. 2007 Nov;35(Pt 5):996-1000. doi: 10.1042/BST0350996. Biochem Soc Trans. 2007. PMID: 17956262 Review.
-
Protein kinase C lambda/iota (PKClambda/iota): a PKC isotype essential for the development of multicellular organisms.J Biochem. 2003 Jan;133(1):9-16. doi: 10.1093/jb/mvg018. J Biochem. 2003. PMID: 12761193 Review.
Cited by
-
Impact of peptide permeation enhancer on tight junctions opening cellular mechanisms.Biochem Biophys Rep. 2022 Oct 27;32:101375. doi: 10.1016/j.bbrep.2022.101375. eCollection 2022 Dec. Biochem Biophys Rep. 2022. PMID: 36324528 Free PMC article.
-
Actin polymerization controls cilia-mediated signaling.J Cell Biol. 2018 Sep 3;217(9):3255-3266. doi: 10.1083/jcb.201703196. Epub 2018 Jun 26. J Cell Biol. 2018. PMID: 29945904 Free PMC article.
-
IRS-1 Functions as a Molecular Scaffold to Coordinate IGF-I/IGFBP-2 Signaling During Osteoblast Differentiation.J Bone Miner Res. 2016 Jun;31(6):1300-14. doi: 10.1002/jbmr.2791. Epub 2016 Feb 20. J Bone Miner Res. 2016. PMID: 26773517 Free PMC article.
-
Integrated Strategies to Gain a Systems-Level View of Dynamic Signaling Networks.Methods Enzymol. 2017;589:133-170. doi: 10.1016/bs.mie.2017.01.016. Epub 2017 Mar 7. Methods Enzymol. 2017. PMID: 28336062 Free PMC article.
-
Reversing the Paradigm: Protein Kinase C as a Tumor Suppressor.Trends Pharmacol Sci. 2017 May;38(5):438-447. doi: 10.1016/j.tips.2017.02.002. Epub 2017 Mar 8. Trends Pharmacol Sci. 2017. PMID: 28283201 Free PMC article. Review.
References
-
- Rosse C., Linch M., Kermorgant S., Cameron A. J., Boeckeler K., Parker P. J. (2010) PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
