

Genetics and genomics of autism spectrum disorder: embracing complexity
- PMID: 26188008
- PMCID: PMC4675826
- DOI: 10.1093/hmg/ddv273
Genetics and genomics of autism spectrum disorder: embracing complexity
Abstract
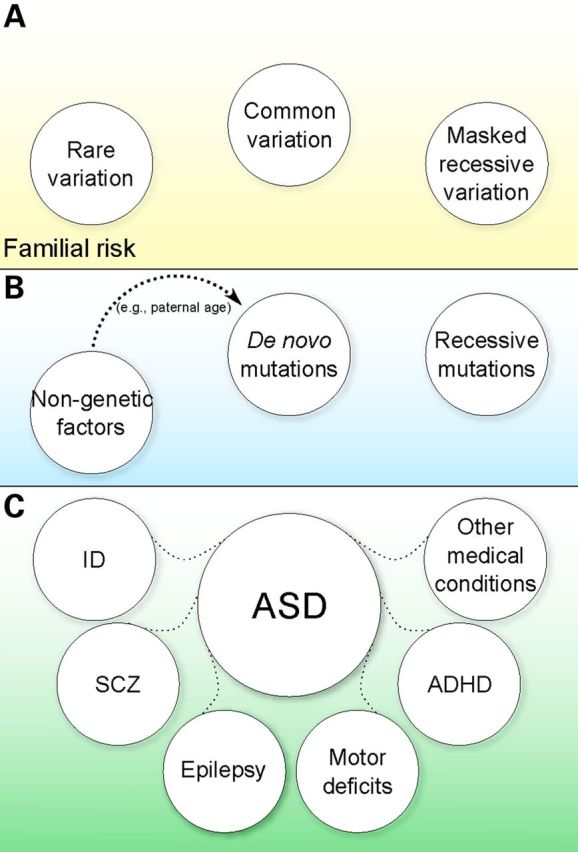
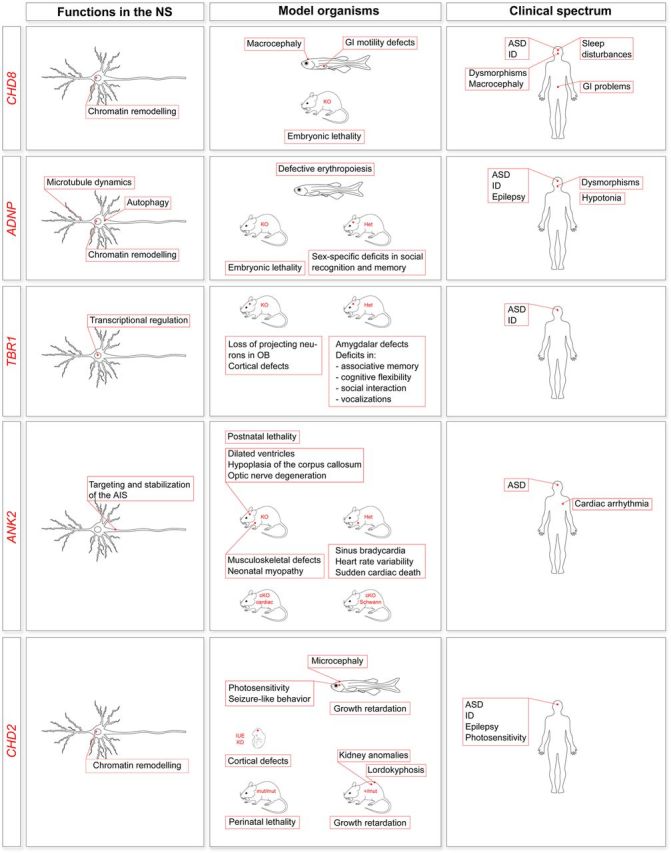
Autism spectrum disorder (ASD) is a neurodevelopmental disorder (NDD) characterized by impairments in social communication and social interaction and the presence of repetitive behaviors and/or restricted interests. ASD has profound etiological and clinical heterogeneity, which has impeded the identification of risk factors and pathophysiological processes underlying the disorder. A constellation of (i) types of genetic variation, (ii) modes of inheritance and (iii) specific genomic loci and genes have all recently been implicated in ASD risk, and these findings are currently being extended with functional analyses in model organisms and genotype-phenotype correlation studies. The overlap of risk loci between ASD and other NDDs raises intriguing questions around the mechanisms of risk. In this review, we will touch upon these aspects of ASD and how they might be addressed.
© The Author 2015. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Szatmari P., Georgiades S., Duku E., Bennett T.A., Bryson S., Fombonne E., Mirenda P., Roberts W., Smith I.M., Vaillancourt T., et al. (2015) Developmental trajectories of symptom severity and adaptive functioning in an inception cohort of preschool children with autism spectrum disorder. JAMA Psychiatry, 72, 276–283. - PubMed
-
- Paul R., Loomis R., Chawarska K. (2014) Adaptive behavior in toddlers under two with autism spectrum disorders. J. Autism Dev. Disord., 44, 264–270. - PubMed
-
- Magiati I., Tay X.W., Howlin P. (2014) Cognitive, language, social and behavioural outcomes in adults with autism spectrum disorders: a systematic review of longitudinal follow-up studies in adulthood. Clin. Psychol. Rev., 34, 73–86. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
