

Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes
- PMID: 26190652
- PMCID: PMC4558313
- DOI: 10.1016/j.cmet.2015.06.010
Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes
Abstract
A number of chronic metabolic pathologies, including obesity, diabetes, cardiovascular disease, asthma, and cancer, cluster together to present the greatest threat to human health. As research in this field has advanced, it has become clear that unresolved metabolic inflammation, organelle dysfunction, and other cellular and metabolic stresses underlie the development of these chronic metabolic diseases. However, the relationship between these systems and pathological mechanisms is poorly understood. Here we discuss the role of cellular Ca(2+) homeostasis as a critical mechanism integrating the myriad of cellular and subcellular dysfunctional networks found in metabolic tissues such as liver and adipose tissue in the context of metabolic disease, particularly in obesity and diabetes.
Copyright © 2015 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors have no conflicts of interest related to the contents of this manuscript.
Figures
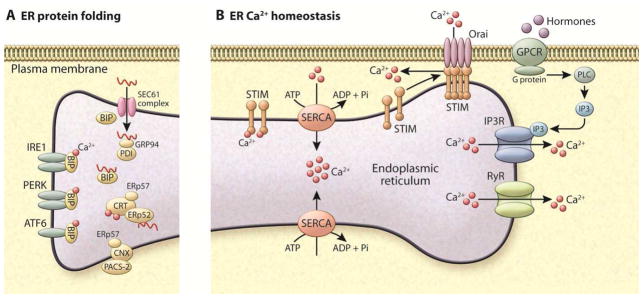
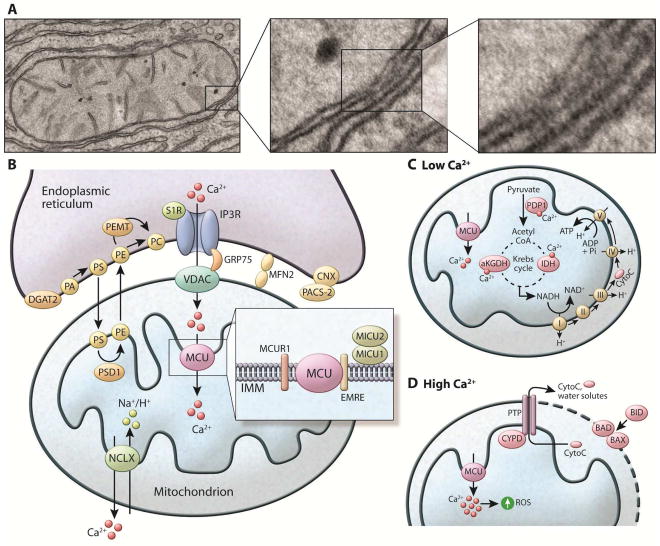
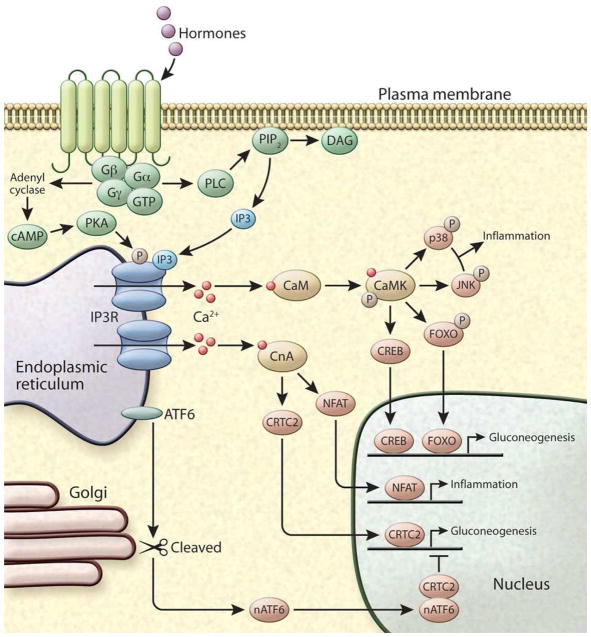
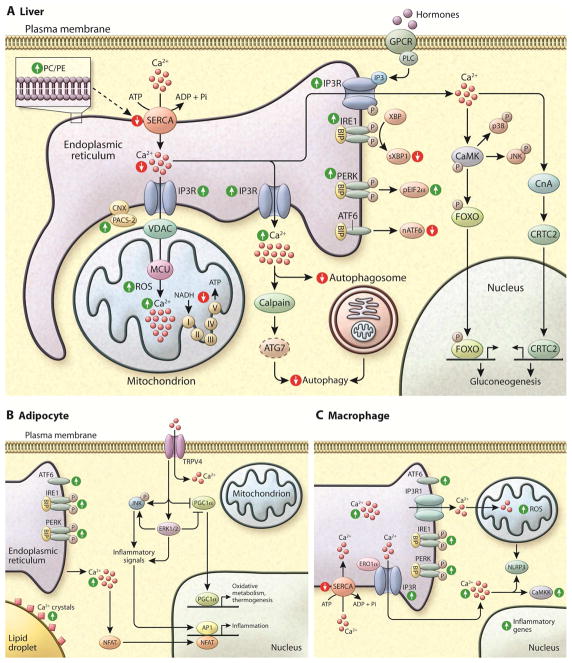
References
-
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. - PubMed
-
- Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nature reviews Endocrinology. 2014;10:24–36. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
