

Whole-genome sequencing provides new insights into the clonal architecture of Barrett's esophagus and esophageal adenocarcinoma
- PMID: 26192915
- PMCID: PMC4556068
- DOI: 10.1038/ng.3357
Whole-genome sequencing provides new insights into the clonal architecture of Barrett's esophagus and esophageal adenocarcinoma
Abstract
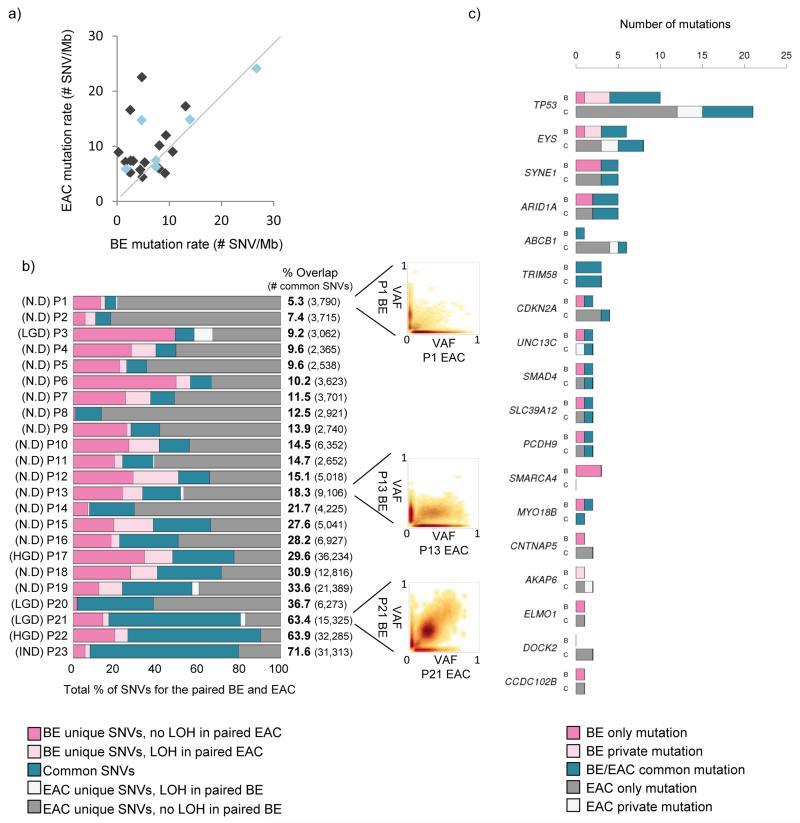
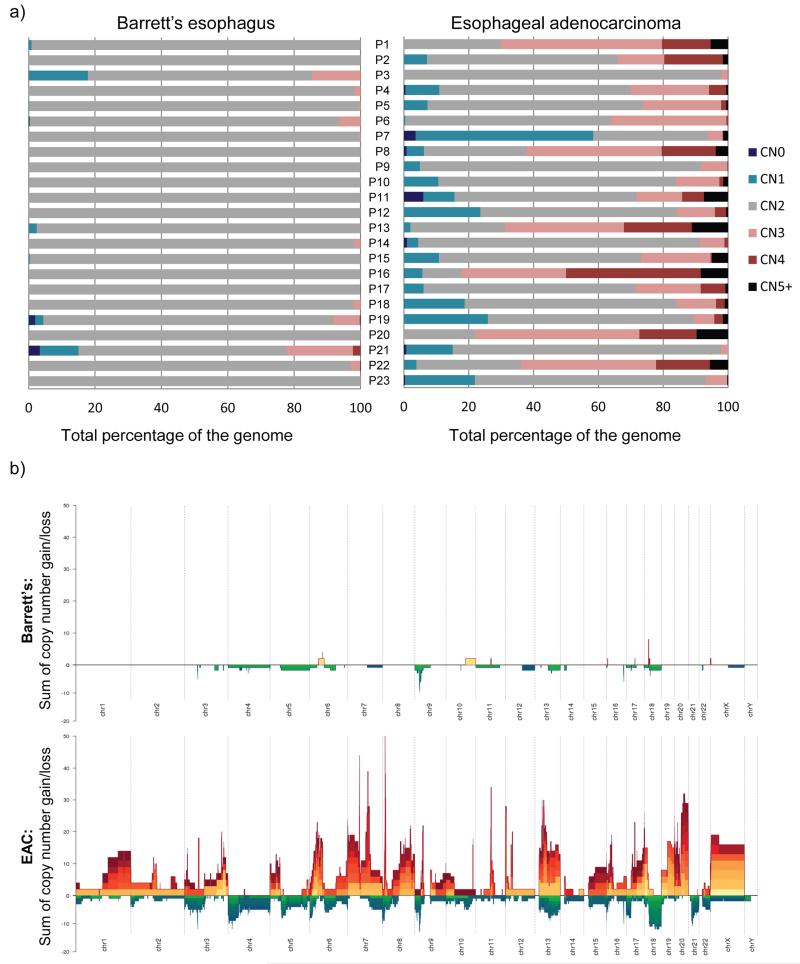
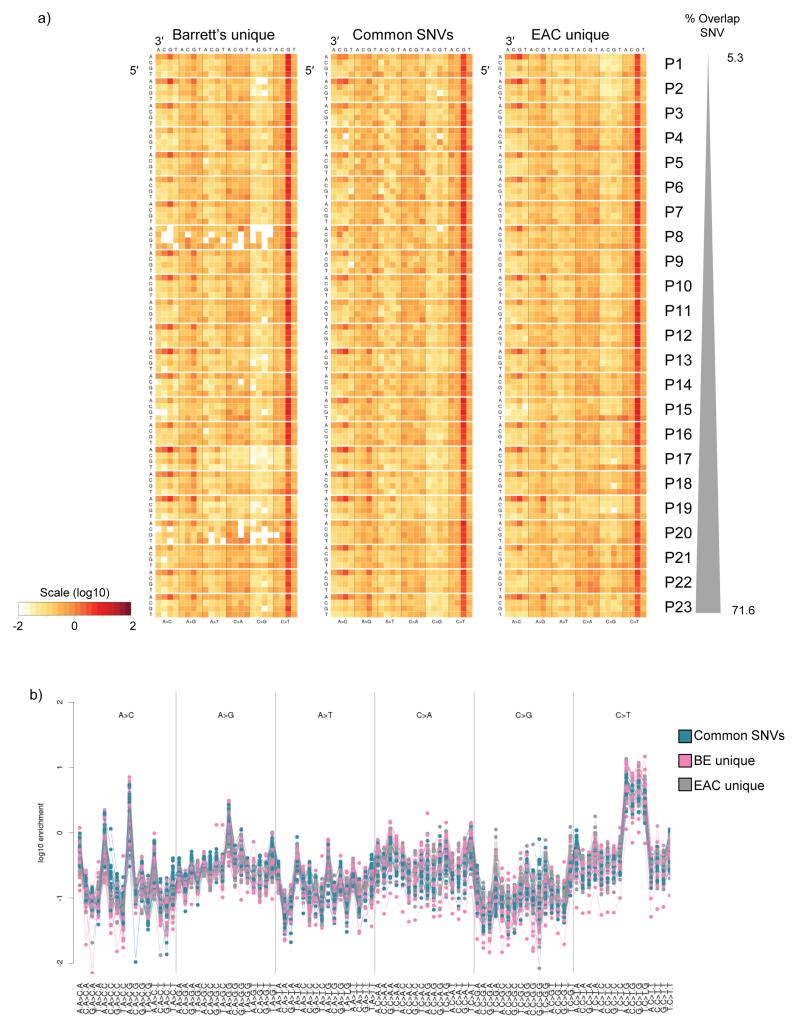
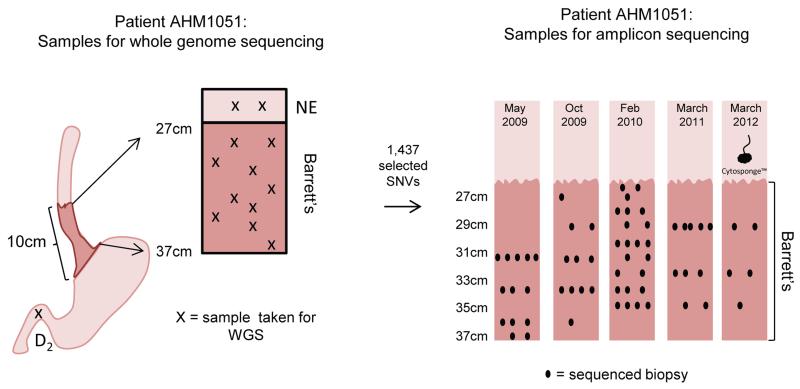
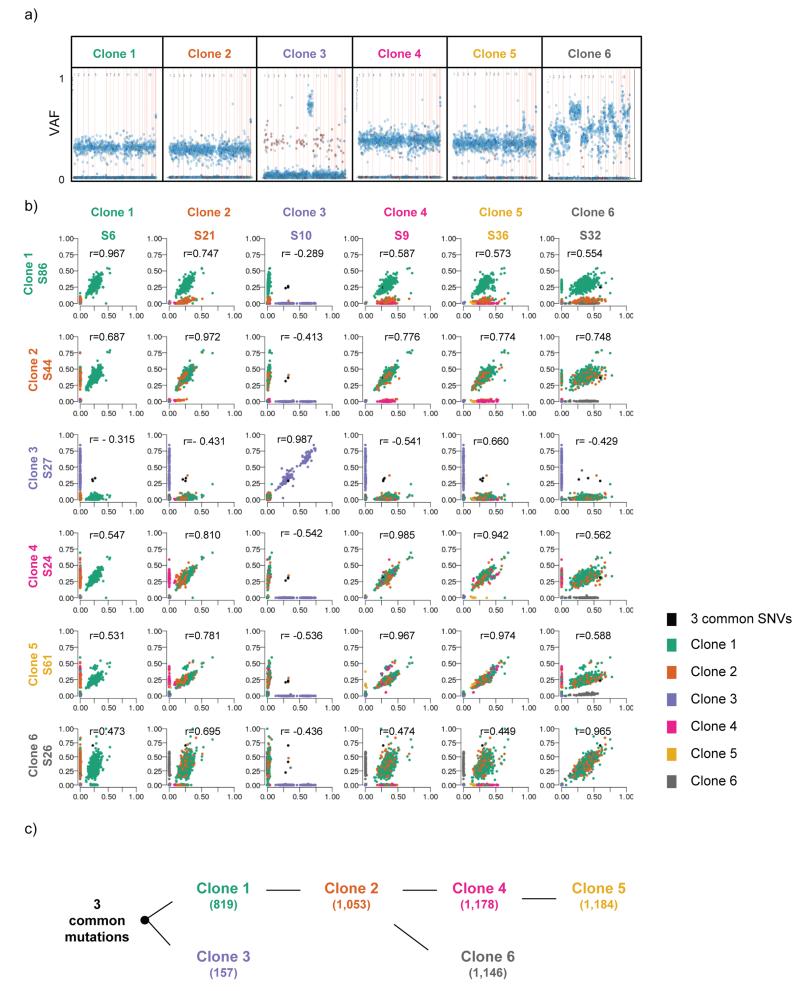
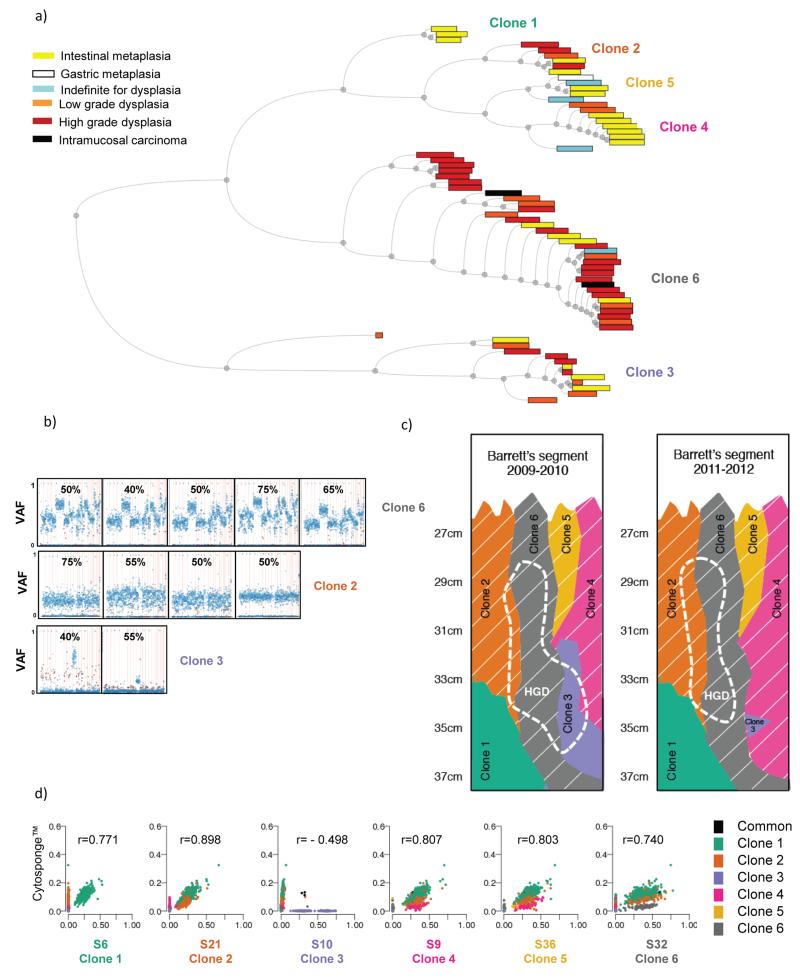
The molecular genetic relationship between esophageal adenocarcinoma (EAC) and its precursor lesion, Barrett's esophagus, is poorly understood. Using whole-genome sequencing on 23 paired Barrett's esophagus and EAC samples, together with one in-depth Barrett's esophagus case study sampled over time and space, we have provided the following new insights: (i) Barrett's esophagus is polyclonal and highly mutated even in the absence of dysplasia; (ii) when cancer develops, copy number increases and heterogeneity persists such that the spectrum of mutations often shows surprisingly little overlap between EAC and adjacent Barrett's esophagus; and (iii) despite differences in specific coding mutations, the mutational context suggests a common causative insult underlying these two conditions. From a clinical perspective, the histopathological assessment of dysplasia appears to be a poor reflection of the molecular disarray within the Barrett's epithelium, and a molecular Cytosponge technique overcomes sampling bias and has the capacity to reflect the entire clonal architecture.
Figures
Comment in
-
Barrett oesophagus: Tracing the genetic pathway to oesophageal cancer.Nat Rev Gastroenterol Hepatol. 2015 Sep;12(9):487. doi: 10.1038/nrgastro.2015.138. Epub 2015 Aug 11. Nat Rev Gastroenterol Hepatol. 2015. PMID: 26260371 No abstract available.
References
Methods-only references
-
- Raczy C, et al. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics. 2013;29:2041–3. - PubMed
-
- Saunders CT, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–7. - PubMed
-
- Gusnanto A, Wood HM, Pawitan Y, Rabbitts P, Berri S. Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics. 2012;28:40–7. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
