

Inferring processes underlying B-cell repertoire diversity
- PMID: 26194757
- PMCID: PMC4528420
- DOI: 10.1098/rstb.2014.0243
Inferring processes underlying B-cell repertoire diversity
Abstract
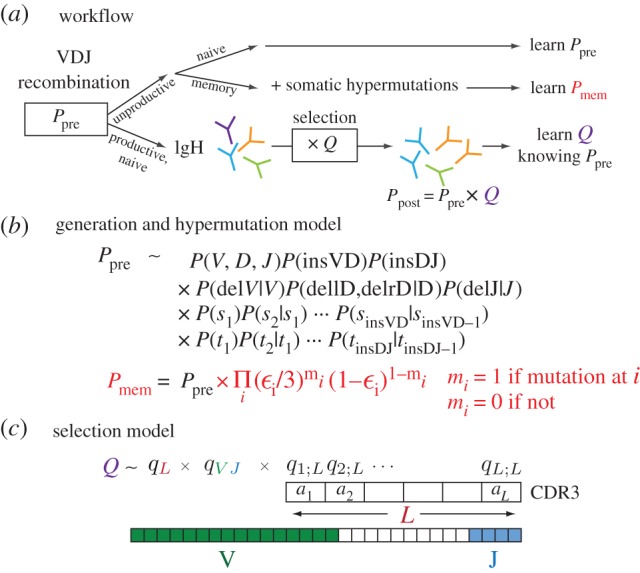
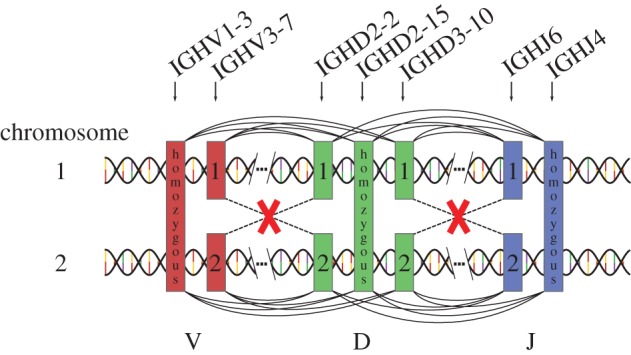
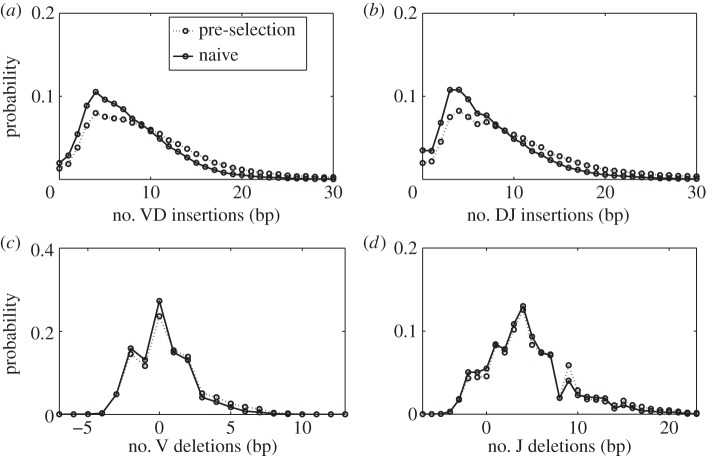
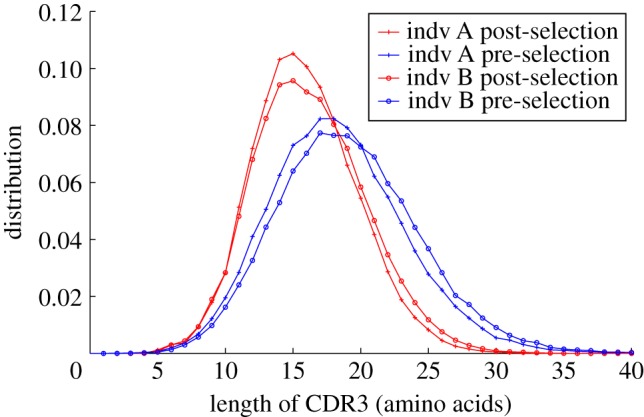
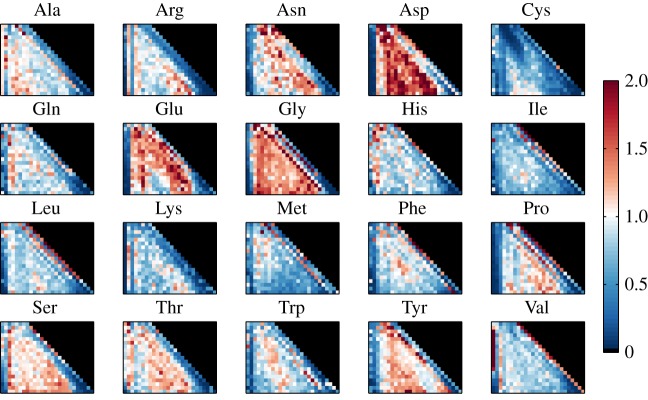
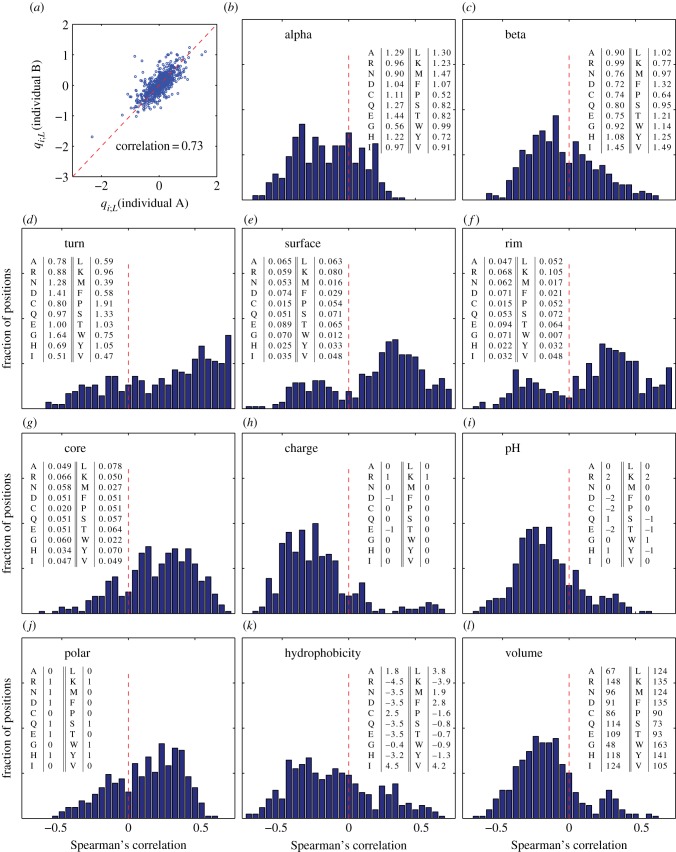
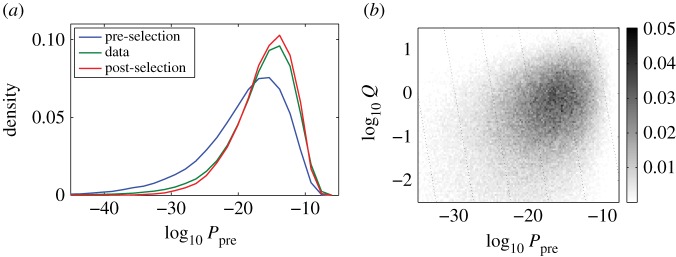
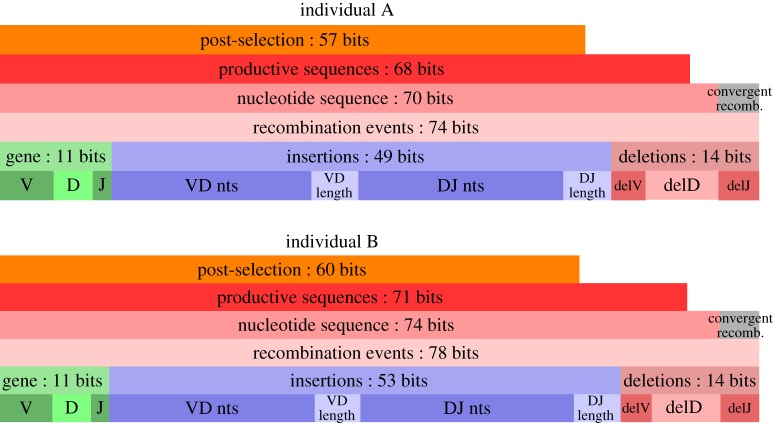
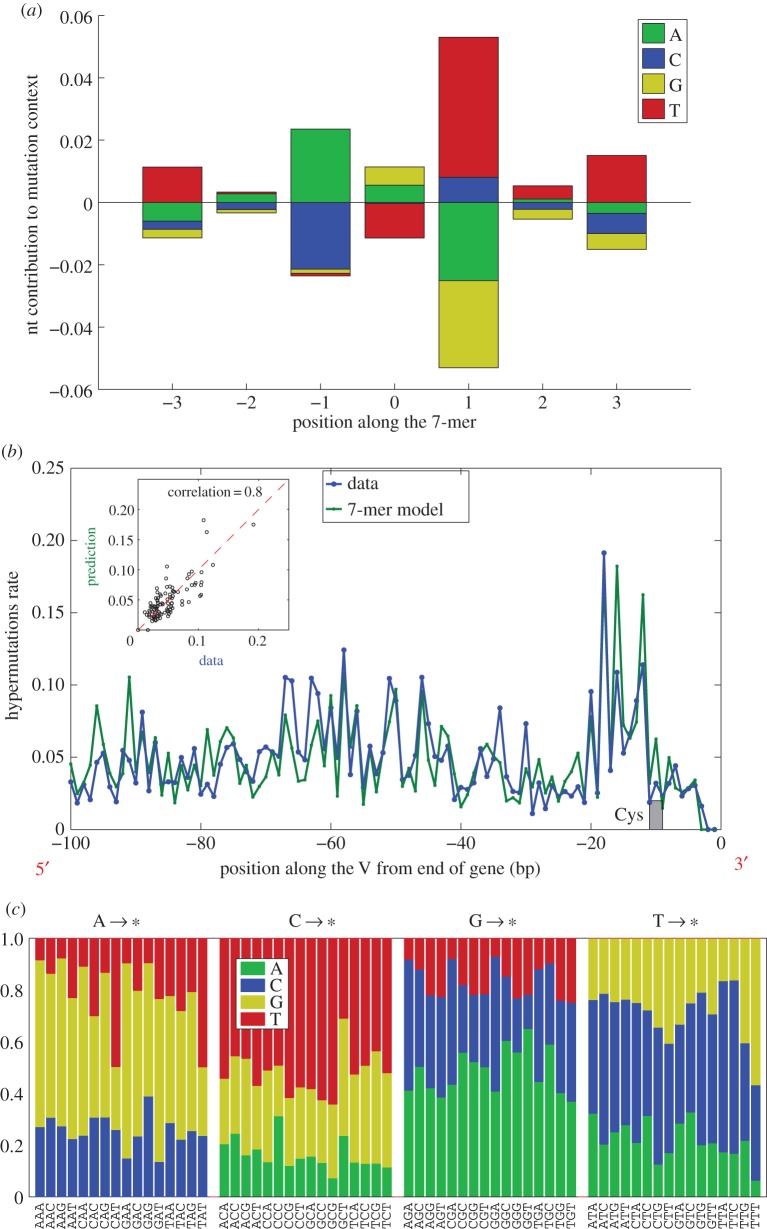
We quantify the VDJ recombination and somatic hypermutation processes in human B cells using probabilistic inference methods on high-throughput DNA sequence repertoires of human B-cell receptor heavy chains. Our analysis captures the statistical properties of the naive repertoire, first after its initial generation via VDJ recombination and then after selection for functionality. We also infer statistical properties of the somatic hypermutation machinery (exclusive of subsequent effects of selection). Our main results are the following: the B-cell repertoire is substantially more diverse than T-cell repertoires, owing to longer junctional insertions; sequences that pass initial selection are distinguished by having a higher probability of being generated in a VDJ recombination event; somatic hypermutations have a non-uniform distribution along the V gene that is well explained by an independent site model for the sequence context around the hypermutation site.
Keywords: B cell; IgH; VDJ recombination; immune repertoire; somatic hypermutations; statistical inference.
Figures
References
-
- Janeway C, Murphy KP, Travers P, Walport M. 2008. Janeway‘s immunobiology. New York, NY: Garland Science.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
