

Strength and precision of neurotransmission at mammalian presynaptic terminals
- PMID: 26194855
- PMCID: PMC4631896
- DOI: 10.2183/pjab.91.305
Strength and precision of neurotransmission at mammalian presynaptic terminals
Abstract
Classically, the basic concept of chemical synaptic transmission was established at the frog neuromuscular junction, and direct intracellular recordings from presynaptic terminals at the squid giant presynaptic terminal have further clarified principles of neurotransmitter release. More recently, whole-cell patch-camp recordings from the calyx of Held in rodent brainstem slices have extended the classical concept to mammalian synapses providing new insights into the mechanisms underlying strength and precision of neurotransmission and developmental changes therein. This review summarizes findings from our laboratory and others on these subjects, mainly at the calyx of Held, with a particular focus on precise, high-fidelity, fast neurotransmission. The mechanisms by which presynaptic terminals acquire strong, precise neurotransmission during postnatal development are also discussed.
Figures
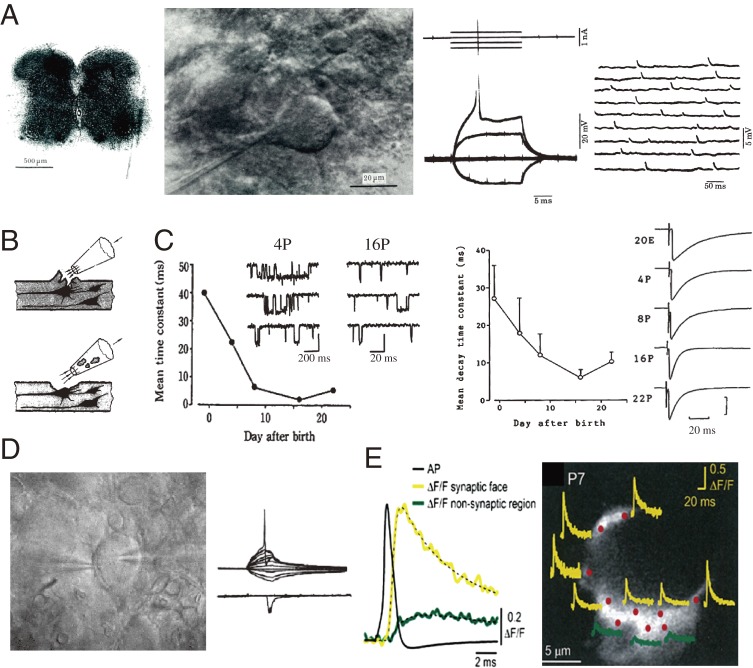
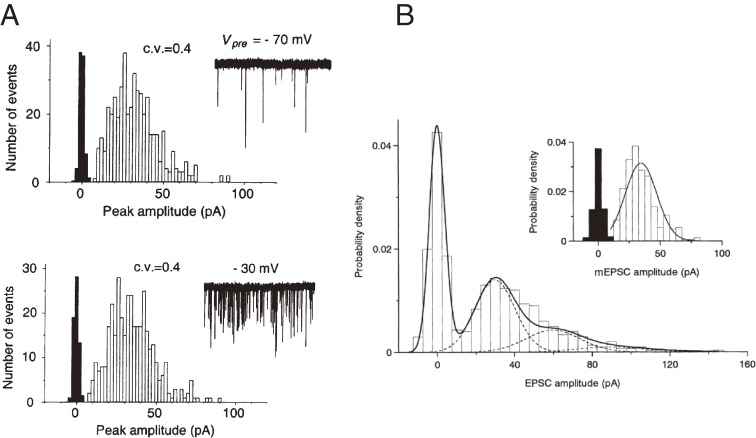
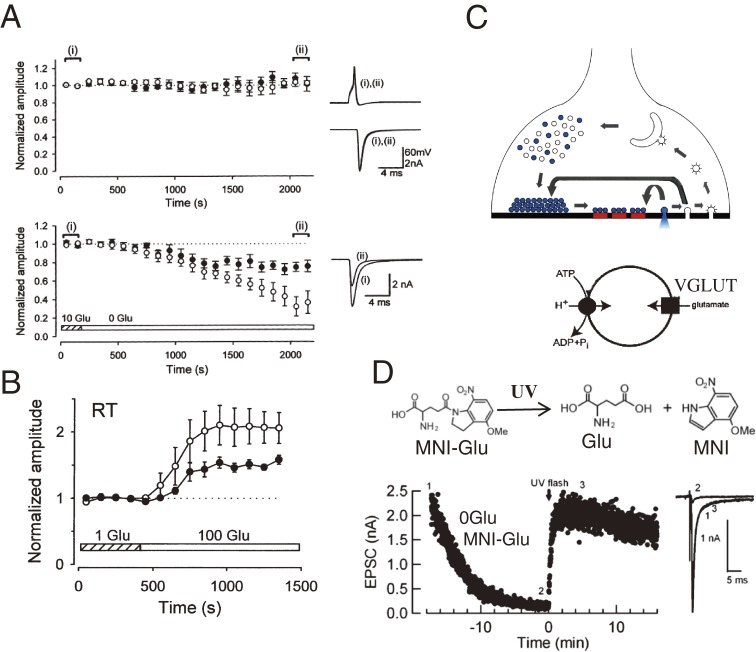
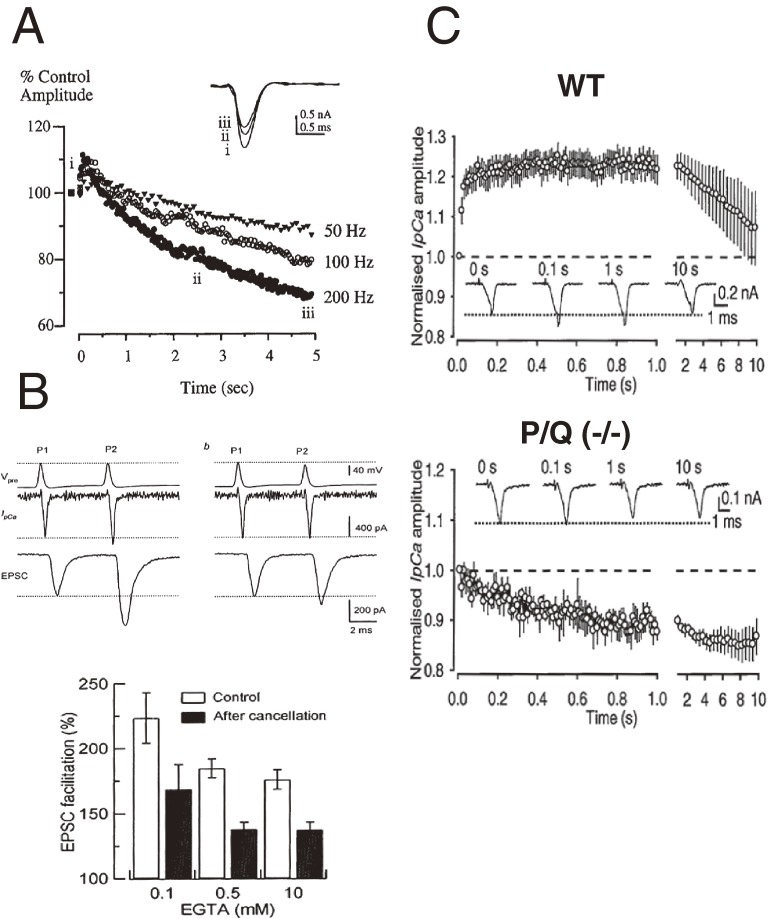
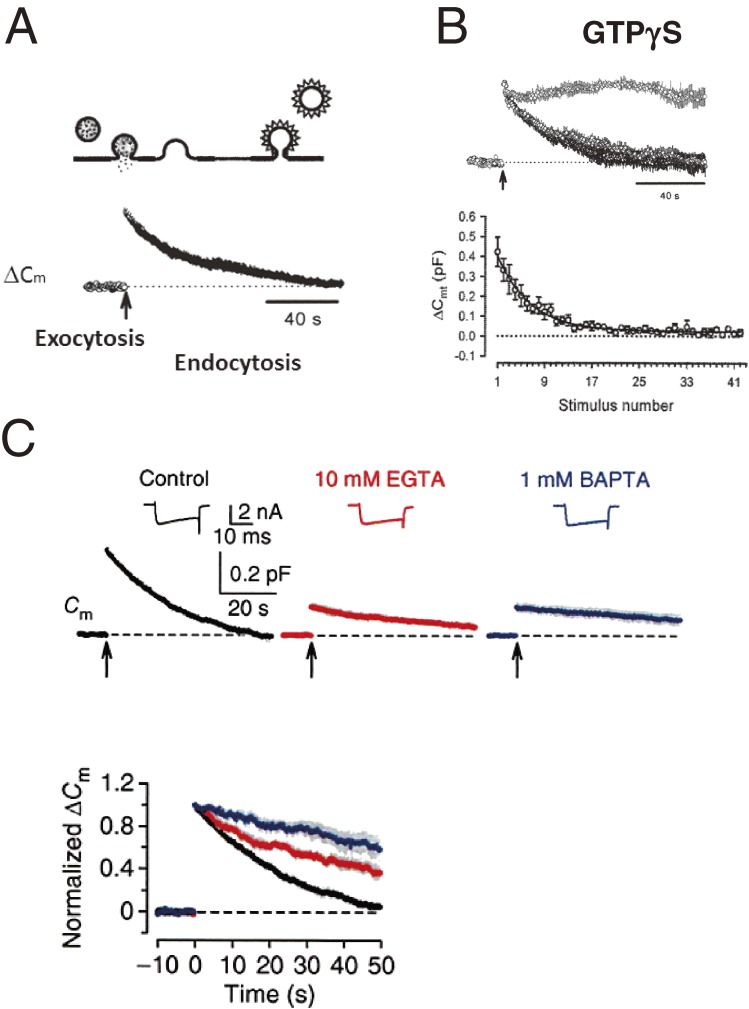
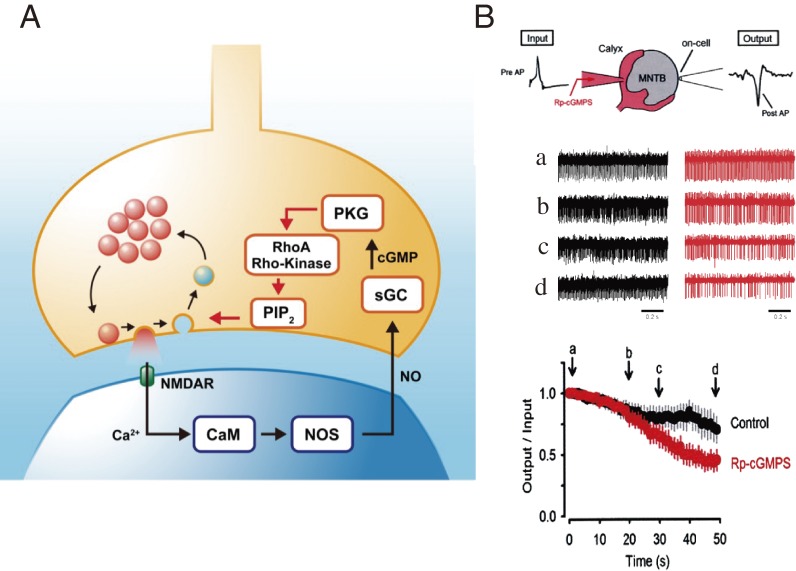
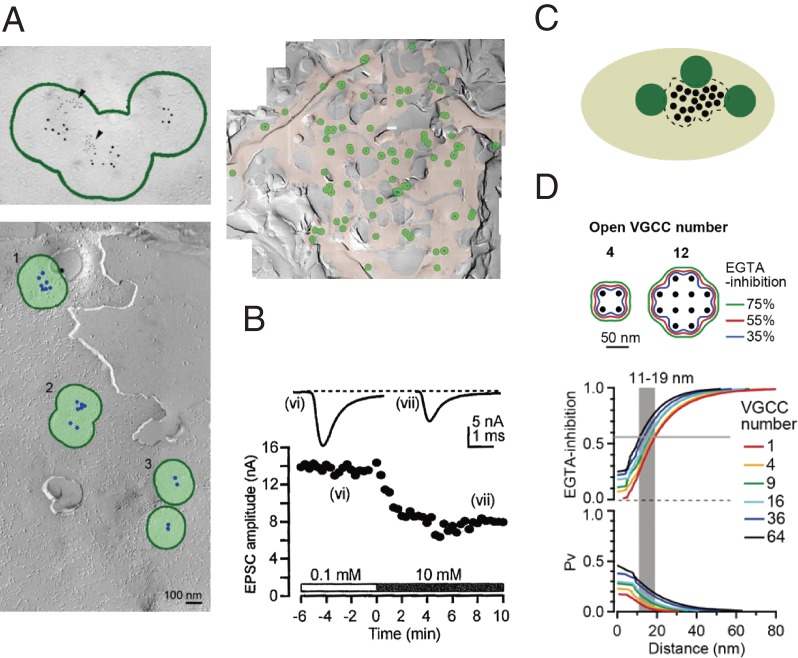
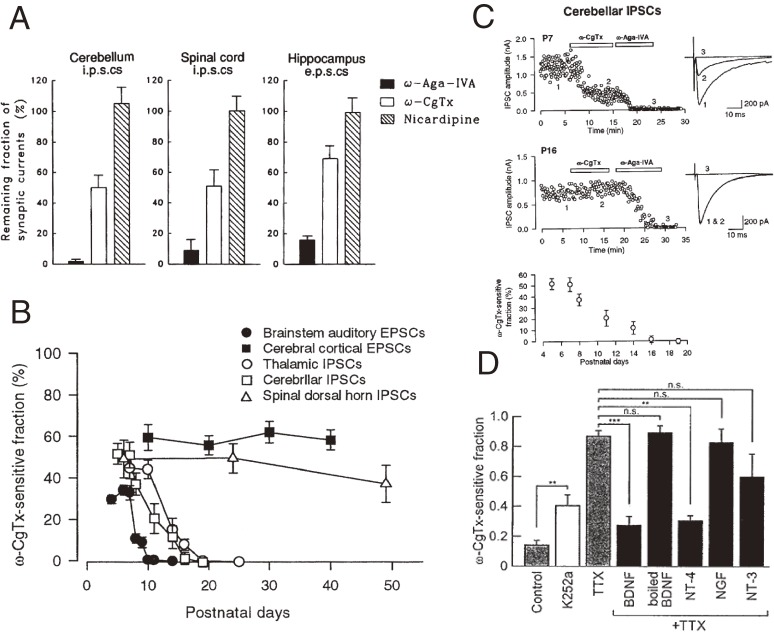
References
-
- Sherrington, C. (1906) The Integrative Action of the Nervous System. Yale Univ. Press, New Haven.
-
- Katz, B. (1969) The Release of Neural Transmitter Substances. The Sherrington Lectures X, Liverpool Univ. Press, Liverpool.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources