

Dual Activation of TRIF and MyD88 Adaptor Proteins by Angiotensin II Evokes Opposing Effects on Pressure, Cardiac Hypertrophy, and Inflammatory Gene Expression
- PMID: 26195481
- PMCID: PMC4537368
- DOI: 10.1161/HYPERTENSIONAHA.115.06011
Dual Activation of TRIF and MyD88 Adaptor Proteins by Angiotensin II Evokes Opposing Effects on Pressure, Cardiac Hypertrophy, and Inflammatory Gene Expression
Abstract
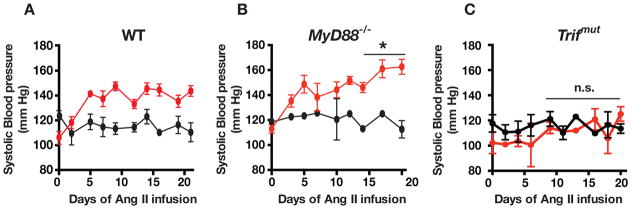
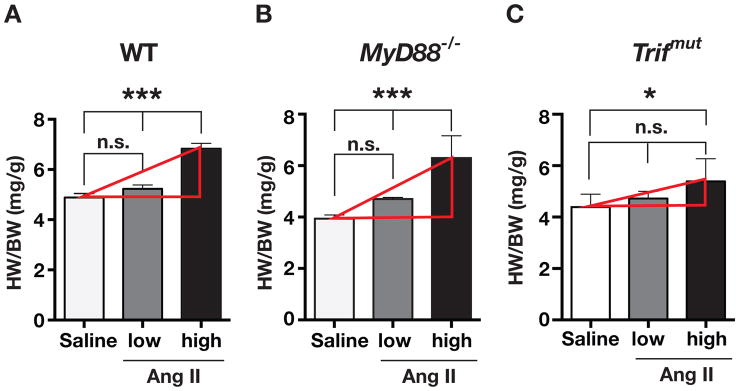
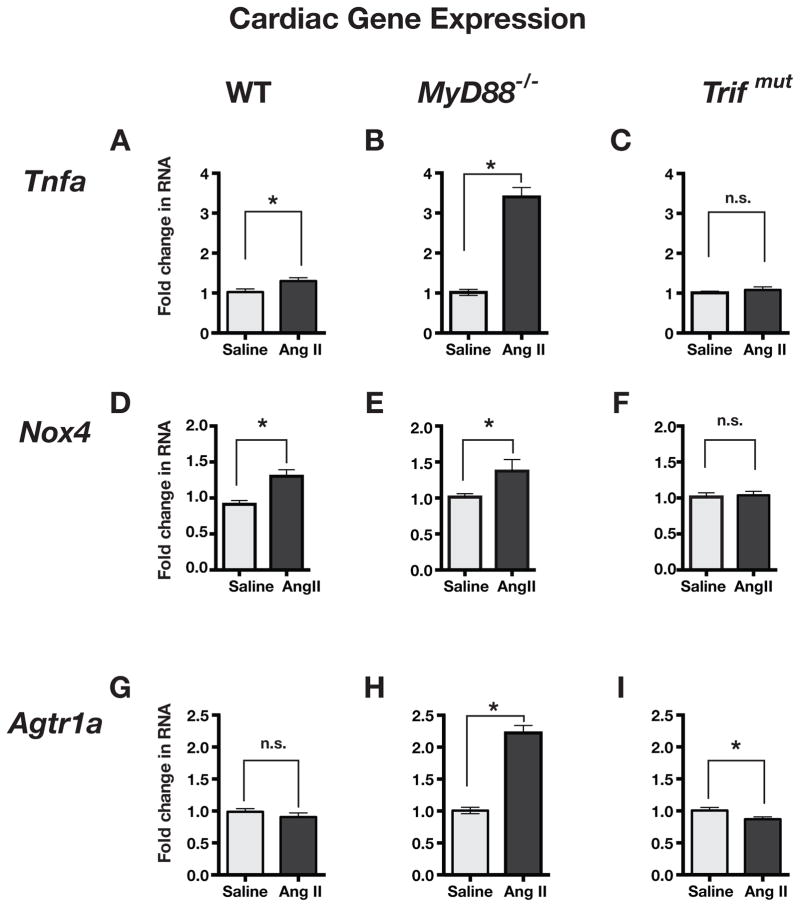
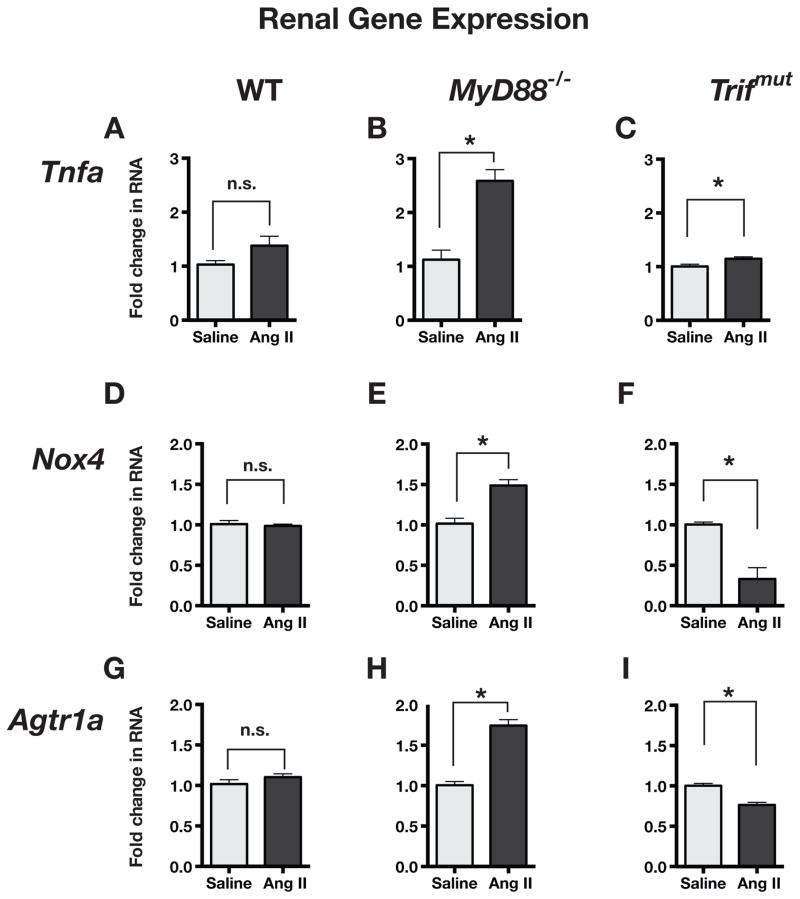
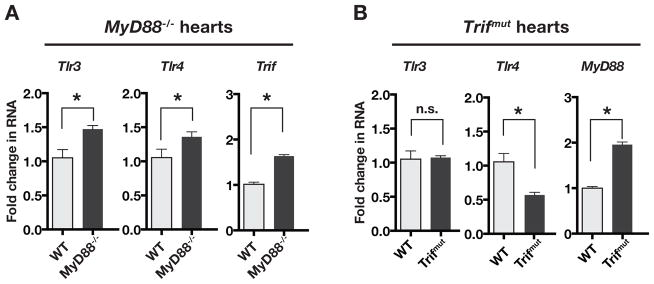
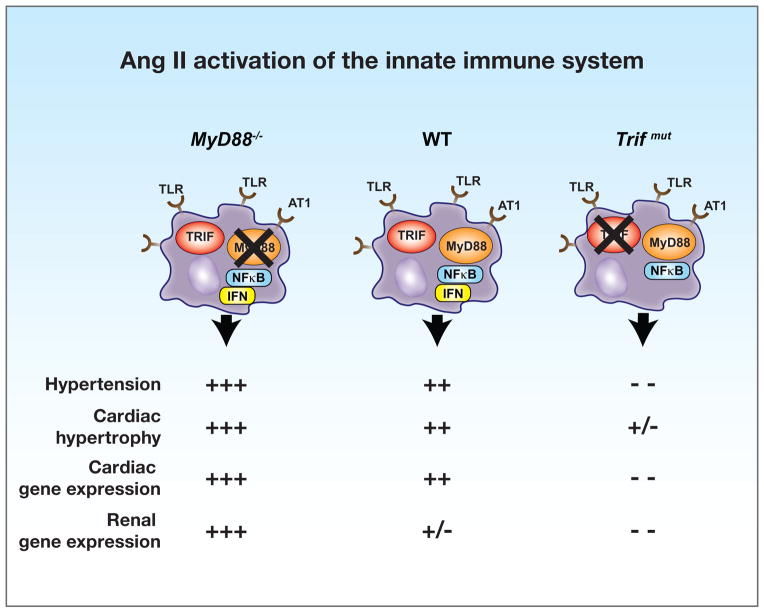
Hypertension is recognized as an immune disorder whereby immune cells play a defining role in the genesis and progression of the disease. The innate immune system and its component toll-like receptors are key determinants of the immunologic outcome through their proinflammatory response. Toll-like receptor-activated signaling pathways use several adaptor proteins of which adaptor proteins myeloid differentiation protein 88 (MyD88) and toll-interleukin receptor domain-containing adaptor protein-inducing interferon-β (TRIF) define 2 major inflammatory pathways. In this study, we compared the contributions of MyD88 and TRIF adaptor proteins to angiotensin II (Ang II)-induced hypertension and cardiac hypertrophy in mice. Deletion of MyD88 did not prevent cardiac hypertrophy and the pressor response to Ang II tended to increase. Moreover, the increase in inflammatory gene expression (Tnfa, Nox4, and Agtr1a) was significantly greater in the heart and kidney of MyD88-deficient mice when compared with wild-type mice. Thus, pathways involving MyD88 may actually restrain the inflammatory responses. However, in mice with nonfunctional TRIF (Trif(mut) mice), Ang II-induced hypertension and cardiac hypertrophy were abrogated, and proinflammatory gene expression in heart and kidneys was unchanged or decreased. Our results indicate that Ang II induces activation of a proinflammatory innate immune response, causing hypertension and cardiac hypertrophy. These effects require functional adaptor protein TRIF-mediated pathways. However, the common MyD88-dependent signaling pathway, which is also activated simultaneously by Ang II, paradoxically exerts a negative regulatory influence on these responses.
Keywords: MyD88 protein; TICAM-1 protein; angiotensin II; gene expression; hypertension; toll-like receptors.
© 2015 American Heart Association, Inc.
Conflict of interest statement
None
Figures
Similar articles
-
Angiotensin II-induced hypertension and cardiac hypertrophy are differentially mediated by TLR3- and TLR4-dependent pathways.Am J Physiol Heart Circ Physiol. 2019 May 1;316(5):H1027-H1038. doi: 10.1152/ajpheart.00697.2018. Epub 2019 Feb 22. Am J Physiol Heart Circ Physiol. 2019. PMID: 30793936 Free PMC article.
-
Aging and contribution of MyD88 and TRIF to expression of TLR pathway-associated genes following stimulation with Porphyromonas gingivalis.J Periodontal Res. 2015 Feb;50(1):89-102. doi: 10.1111/jre.12185. Epub 2014 May 24. J Periodontal Res. 2015. PMID: 24862405 Free PMC article.
-
Regulation of lipopolysaccharide-inducible genes by MyD88 and Toll/IL-1 domain containing adaptor inducing IFN-beta.Biochem Biophys Res Commun. 2005 Mar 11;328(2):383-92. doi: 10.1016/j.bbrc.2004.12.184. Biochem Biophys Res Commun. 2005. PMID: 15694359
-
Role of Adaptor Protein Myeloid Differentiation 88 (MyD88) in Post-Subarachnoid Hemorrhage Inflammation: A Systematic Review.Int J Mol Sci. 2021 Apr 18;22(8):4185. doi: 10.3390/ijms22084185. Int J Mol Sci. 2021. PMID: 33919485 Free PMC article.
-
Dual functional roles of the MyD88 signaling in colorectal cancer development.Biomed Pharmacother. 2018 Nov;107:177-184. doi: 10.1016/j.biopha.2018.07.139. Epub 2018 Aug 4. Biomed Pharmacother. 2018. PMID: 30086464 Review.
Cited by
-
Protease-Activated Receptor 1 Contributes to Angiotensin II-Induced Cardiovascular Remodeling and Inflammation.Cardiology. 2017;136(4):258-268. doi: 10.1159/000452269. Epub 2016 Nov 24. Cardiology. 2017. PMID: 27880950 Free PMC article.
-
Key Player in Cardiac Hypertrophy, Emphasizing the Role of Toll-Like Receptor 4.Front Cardiovasc Med. 2020 Nov 26;7:579036. doi: 10.3389/fcvm.2020.579036. eCollection 2020. Front Cardiovasc Med. 2020. PMID: 33324685 Free PMC article. Review.
-
Systemic leukotriene B4 receptor antagonism lowers arterial blood pressure and improves autonomic function in the spontaneously hypertensive rat.J Physiol. 2016 Oct 15;594(20):5975-5989. doi: 10.1113/JP272065. Epub 2016 Jul 8. J Physiol. 2016. PMID: 27230966 Free PMC article.
-
Targeting cytokine signaling in salt-sensitive hypertension.Am J Physiol Renal Physiol. 2016 Dec 1;311(6):F1153-F1158. doi: 10.1152/ajprenal.00273.2016. Epub 2016 Aug 24. Am J Physiol Renal Physiol. 2016. PMID: 27558557 Free PMC article. Review.
-
Choline ameliorates cardiovascular damage by improving vagal activity and inhibiting the inflammatory response in spontaneously hypertensive rats.Sci Rep. 2017 Feb 22;7:42553. doi: 10.1038/srep42553. Sci Rep. 2017. PMID: 28225018 Free PMC article.
References
-
- Rodriguez-Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol. 2014;10:56–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
